Inhibicí kinázy, která je součástí signální dráhy v buňce, se ovlivní transkripce nebo translace genů a tím se může zastavit buněčný růst, proliferace nebo metabolismus buňky. Účinnost kinázových inhibitorů je ovlivněna několika faktory. Signální dráhy v buňkách jsou velmi komplexní a vyřazení jednoho proteinu může vést k využívání alternativní signální dráhy. Některé molekuly ovšem inhibicí hned několika proteinů působí na více místech a omezují využívání alternativních drah. Cílový protein také může být mutován takovým způsobem, že na něj molekula léčiva nepůsobí. K takovému vzniku rezistence může dojít i v průběhu léčby anebo při relapsu.
Molekulárně genetické vyšetření nádorové tkáně může poskytnout informaci o tom, jaké signální dráhy nádorová buňka využívá, jsou-li v kinázách přítomny konkrétní mutace a jaká léčba by tak mohla být účinná. V celé řadě případů je toto vyšetření podmínkou úhrady z prostředků veřejného zdravotního pojištění (toto je uvedeno v předchozích kapitolách u jednotlivých diagnóz). V případě vzácných nádorů nebo nádorů neznámého origa může molekulárně genetické vyšetření pomoci indikovat účinnou léčbu.
Kapitola se věnuje problematice farmakokinetických interakcí cílených léčiv, tzv. malých molekul, z nichž mnohé jsou metabolizovány dominantně jednou formou cytochromu P450 (CYP), popř. jsou inhibitory forem CYP. Z toho plyne nezanedbatelné riziko farmakokinetických interakcí, ať už směrem k metabolismu těchto léčiv, z nichž mnohá mají nepříliš široké terapeutické rozmezí anebo opačným směrem, kdy cílená léčba může zásadně změnit koncentrace souběžné medikace. Vedle cytochromu P450 často dochází k farmakokinetickým interakcím i na transportních proteinech jako jsou P-glykoprotein, BCRP nebo konjugačních enzymech, především uridin glukuronyl transferáza (UGT). Interakce farmakodynamické, ke kterým také často dochází (např. sumace nebo potenciace orgánové toxicity, ovlivnění QT intervalu apod.), jsou příliš heterogenní na to, aby je bylo možno přehledně prezentovat; je tedy třeba je posuzovat a řešit individuálně, popř. ve spolupráci s klinickým farmaceutem.
Přehled v Česku registrovaných léčiv skupiny tzv. cílené terapie malými molekulami a jejich cílových struktur uvádí následující tabulka.
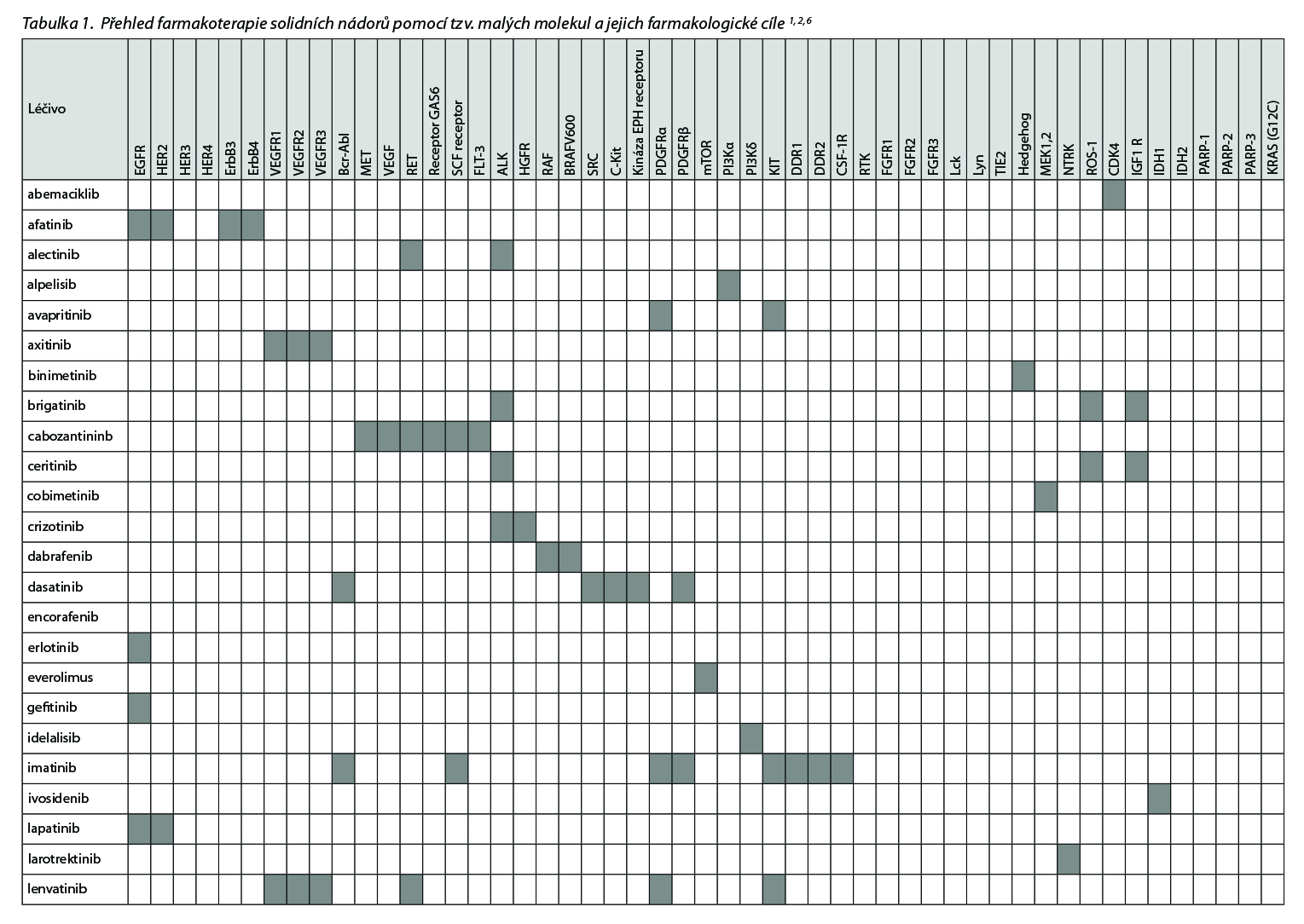
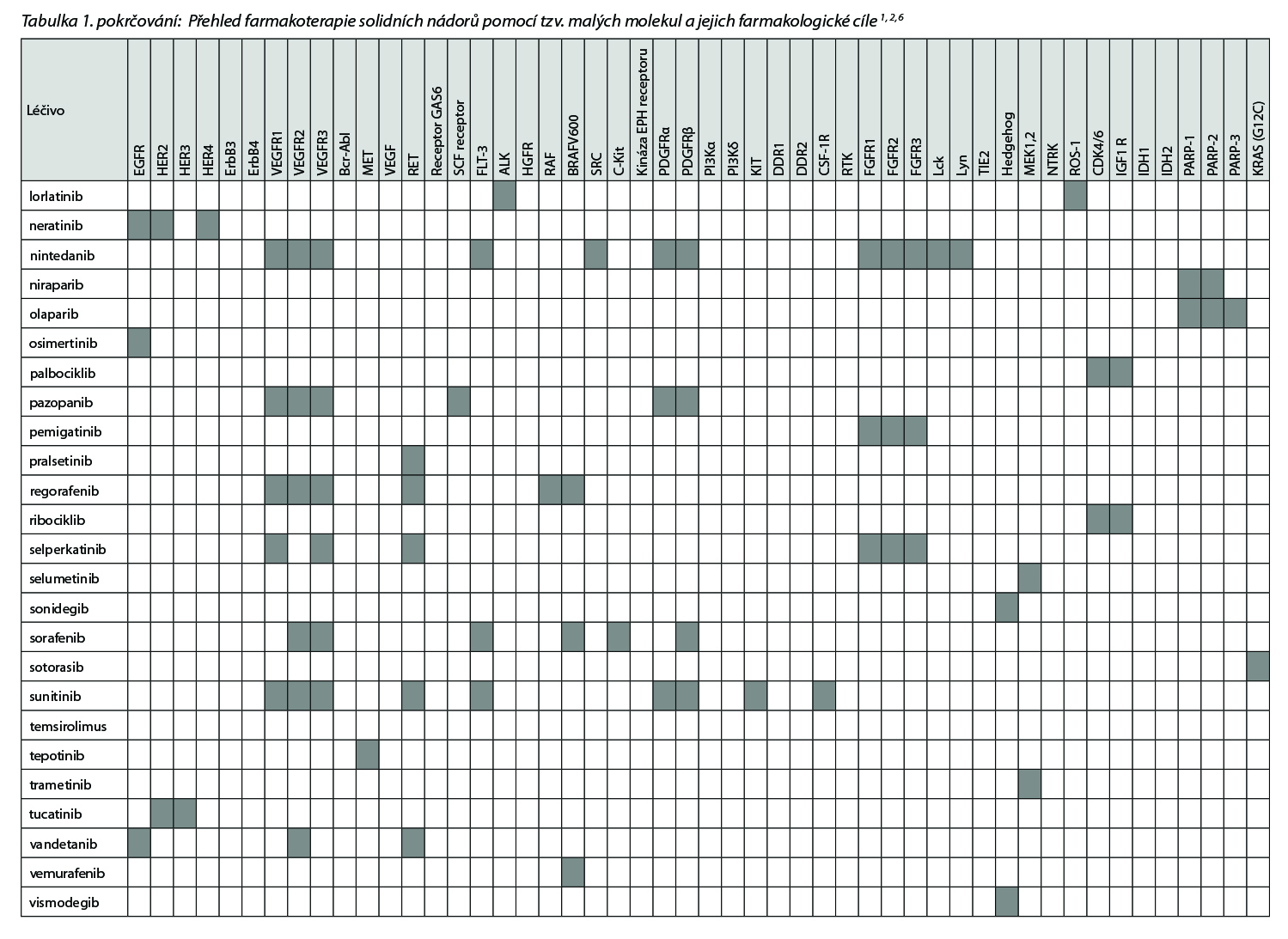
Farmakokinetické interakce
K farmakokinetickým interakcím dochází obecně na úrovni absorpce, distribuce, biotransformace nebo vylučování léčiv. Tyto interakce mohou být velmi závažné, protože v některých případech může dojít až k několikanásobnému zvýšení plazmatické koncentrace (nebo snížení, dle typu konkrétní interakce) a tím i k nežádoucím účinkům nebo selhání léčby – a to jak kinázovými inhibitory, tak i ostatní souběžně podávané medikace.
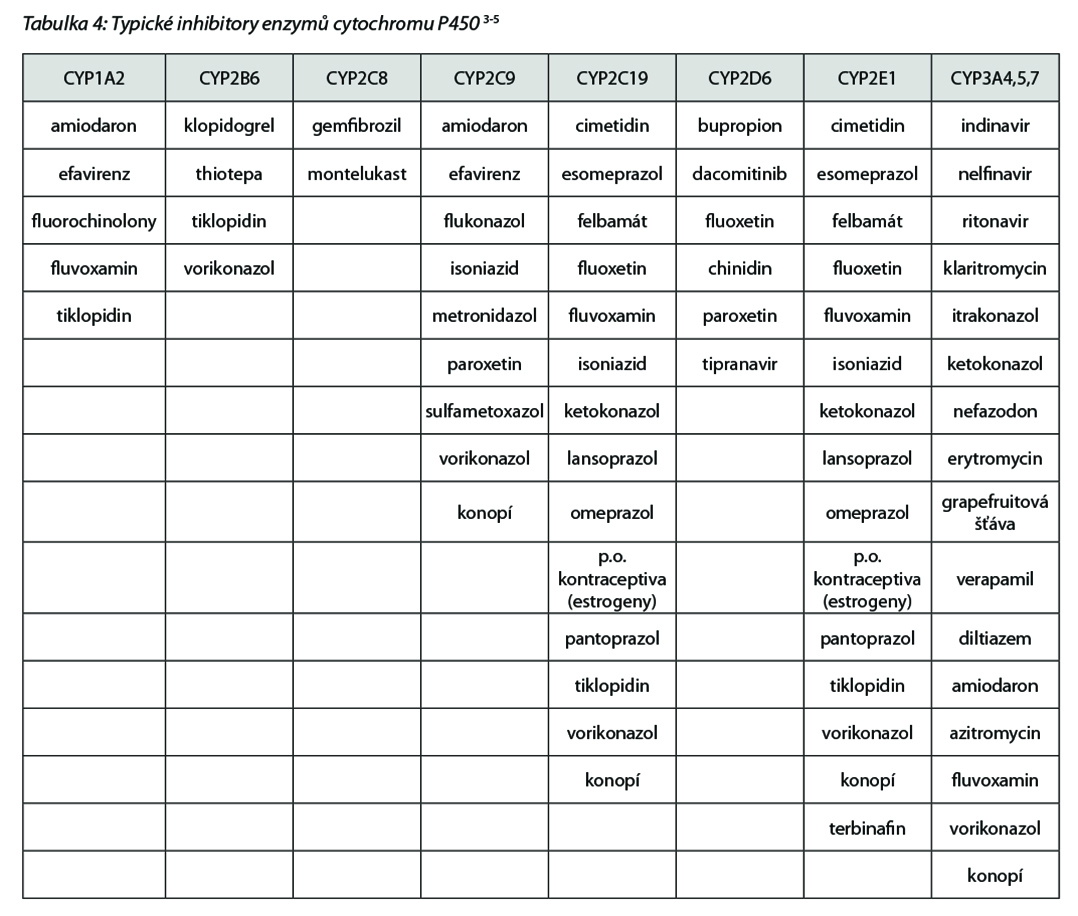
Nejčetnější jsou interakce na úrovni cytochromu P450 (CYP). Všechna léčiva mohou být substráty, induktory a inhibitory izoforem CYP. Jedno léčivo může být v určitých poměrech metabolizováno současně několika izoformami a pro každou z nich může být tato molekula nezávisle na sobě jako substrát, inhibitor nebo induktor (např. omeprazol je substrátem
CYP2C19 a CYP3A4 a současně inhibitorem CYP2C19, diltiazem je substrátem CYP2C9 a zároveň inhibitorem CYP3A4 apod.). V tabulce 2 jsou uvedeny enzymy a transportéry, které metabolizují jednotlivé látky.
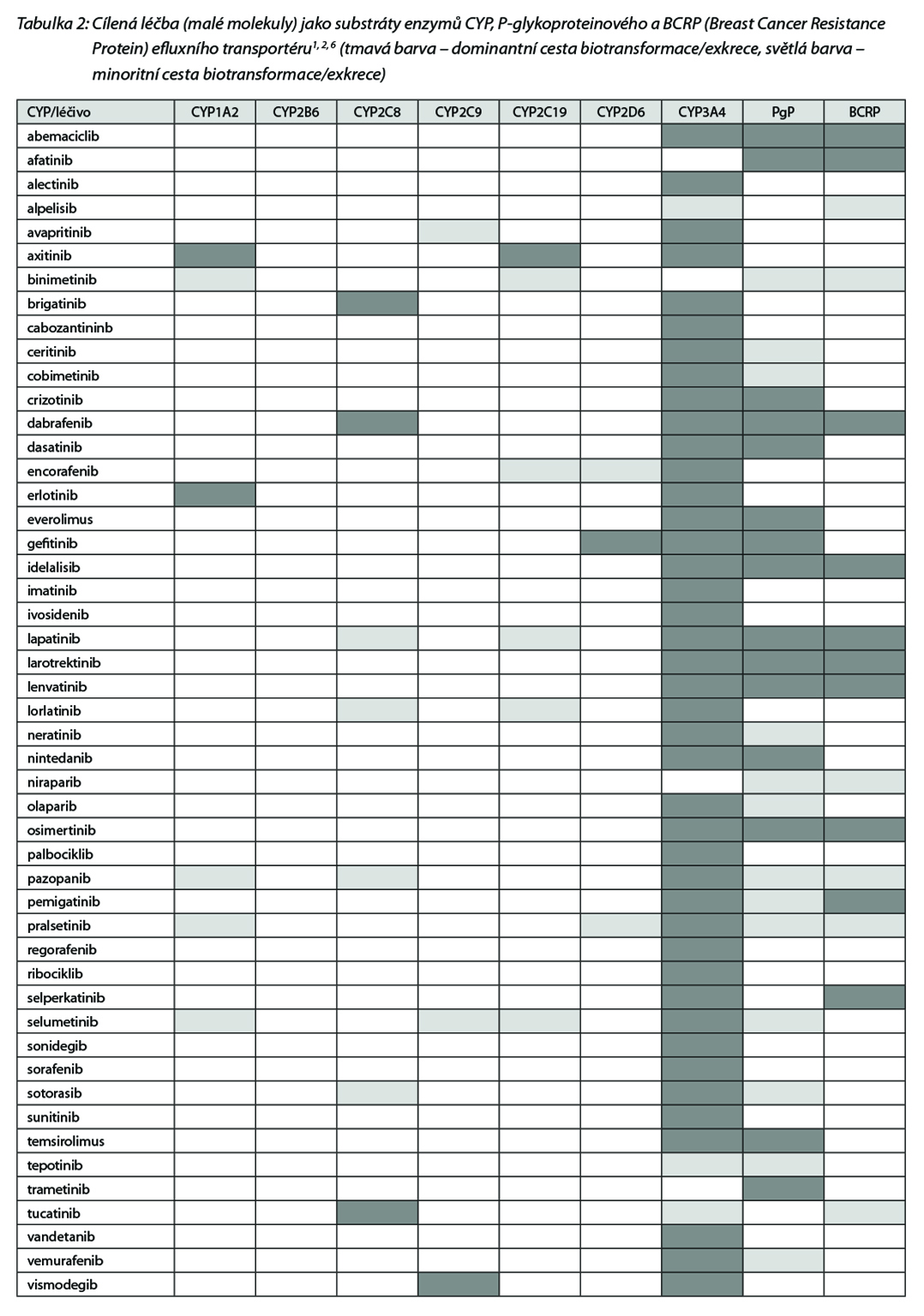
V případě indukce vlivem opakovaného podání jednoho léčiva (např. karbamazepin, rifampicin, typické induktory viz níže v tab. 3) dojde k přechodnému zvýšení exprese enzymu. Tím jsou substráty metabolizovány rychleji a plazmatické koncentrace jsou pak nižší (např. sunitinib + dexametazon/rifampicin/třezalka tečkovaná). To pak může projevit nižší intenzitou efektu a/nebo selháním terapie.
Zatímco k indukci dojde obvykle až při opakovaném podání, v řádu přinejmenším několika dnů, v případě inhibice může dojít k interakci okamžitě, i vlivem jednorázového předchozího či současného podání inhibitoru. Důsledky jsou opačné než v případě indukce – vlivem inhibitoru (např. fluoxetin, ketokonazol, tab. 4 níže) dochází ke zpomalení biotransformace, zvýšení plazmatické koncentrace a plochy pod koncentrační křivkou (AUC), což může mít důsledek jednak ve zvýšeném účinku, ale také zvýšení frekvence a/nebo intenzity nežádoucích účinků jak cílené protinádorové léčby, tak i ostatní souběžně podávané medikace (klaritromycin, ritonavir a sunitinib).
Výše uvedené platí v případě, že podáváme aktivní formu léčiva, tedy nikoli proléčivo (pro-drug). U proléčiv naopak dochází k opačným efektům: inhibitory biotransformace sníží koncentrace aktivní formy a tím mohou snížit i velikost efektu, naopak induktory mohou zrychlit aktivaci léčiva a tím zvýšit plazmatické koncentrace a potažmo velikost efektu a toxicitu. V případě, že podáváme aktivní molekulu a ta má navíc i podobně aktivní metabolit (např. sunitinib), je situace složitější a zasluhuje si komplexní posouzení případného ovlivnění všech hlavních biotransformačních drah.
Vliv cílené léčby (malých molekul) na jiná léčiva
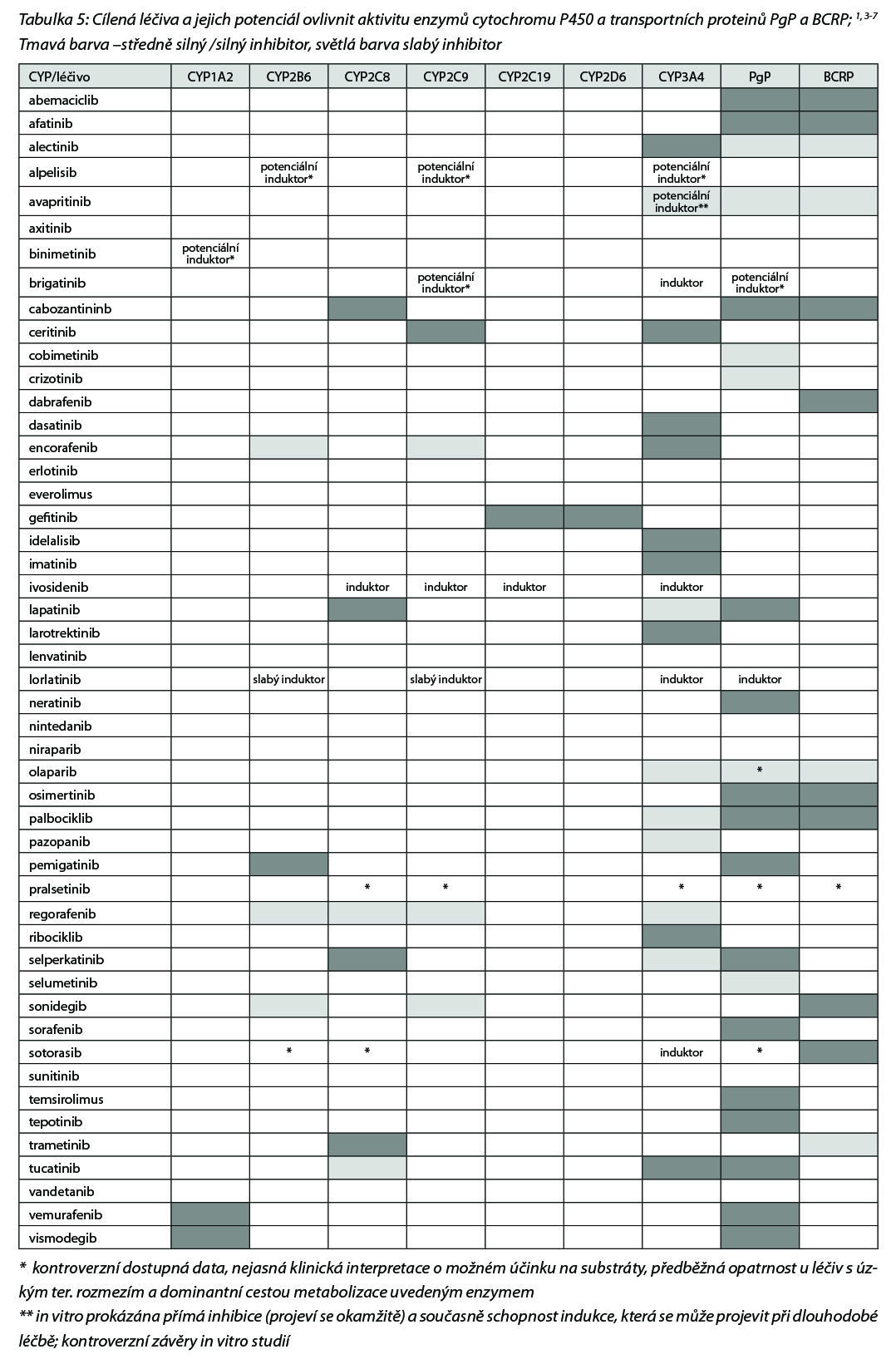
Nezanedbatelný je také potenciální vliv malých molekul na metabolismus jiných léčiv, protože mnohé kinázové inhibitory jsou klinicky významnými inhibitory biotransformačních enzymů a transportních proteinů, Tato inhibice pak může vyústit v toxicitu nebo četné nežádoucí účinky především u léčiv s úzkým terapeutickým oknem. Přehled léčiv (cílené léčby, malé molekuly) jako inhibitorů enzymů CYP a transportních proteinů BCRP a PgP uvádí Tabulka 5.
Vztah expozice a účinku, terapeutické referenční rozmezí
U většiny zmíněných léčiv je dobře znám vztah mezi účinkem, potažmo toxicitou a plazmatickou koncentrací. Jako měřítka efektu se používá nejčastěji TTF, OS a PFS.
Měřítkem expozice je pak plocha pod koncentrační křivkou (AUC), která je často extrapolovaná z údolní koncentrace léčiva (tj. minimální koncentrace před podáním opakované dávky léčiva). Častěji se proto přistupuje právě k monitoringu této hladiny (tzv. trough level, Ctrough). V případě dobře známé farmakokinetiky konkrétního léčiva lze v některých případech také extrapolovat Ctrough z hladiny odebrané v jiný čas, nejlépe však alespoň 3 hodiny po podání.
Přinejmenším u některých kinázových inhibitorů (např. imatinib, sunitinib, pazopanib) je dostatečná evidence pro vhodnost terapeutického monitorování plazmatických hladin. Byly publikovány studie sledující proveditelnost a benefit intervenčního TDM, na jehož základě docházelo k úpravě dávky u pacientů se solidními nádory.7
Literatura:
1. UpToDate: UpToDate, Waltham, MA.; 2024.
2. InfoPharm. AISLP; 2024.
3. Anzenbacher P, Anzenbacherová E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. May 2001;58(5-6):737-47.
4. Flockhart D. CYTOCHROME P450 DRUG-INTERACTION TABLE; 2008.
5. Nelson DR. Human P450 table; 2003.
6. EMA-European Medicines Agency; SmPC přípravků
7. Lankheet NA, Kloth JS, Gadellaa-van Hooijdonk CG et al. Pharmacokinetically guided sunitinib dosing: a feasibility study in patients with advanced solid tumours. Br J Cancer. 2014; 110; 2441-2449.


