Obsah:
Zhoubné nádory nitrooční
Retinoblastom
Retinoblastom (RB) je maligní nádor sítnice oka. Představuje 1 až 2% ze všech dětských nádorů a je nejčastějším očním nádorem u dětí. Ročně je v ČR diagnostikováno kolem 10 nových případů, v celém světě přibližně 5000 nově zjištěných případů RB. Retinoblastom vyrůstá z buněk sítnice, které jsou odvozené z embryonální neurální lišty (neuroepitelu). To vysvětluje jeho částečnou podobnost s dalším (mnohem častějším) embryonálním maligním nádorem dětského věku - neuroblastomem.
Onemocnění retinoblastomem může být klasifikováno ze tří hledisek: sporadický nebo familiární typ; jednostranné či oboustranné postižení očí; a dědičný (germinální mutace) nebo nedědičný typ (somatická mutace).
Epidemiologie
V ekonomicky vyspělých zemích se retinoblastom vyskytuje s četností 1 nádor na 15000 - 18000 novorozenců za rok. V populaci do 15 let je incidence 5/100 000. V rozvojových zemích Latinské Ameriky, Afriky a Asie je výskyt onemocnění vyšší. 40-50% nemocných má formu hereditární, 50-60% sporadickou. Odlišení formy onemocnění je zcela zásadní pro prognózu, správnou léčbu a vhodné poradenství rodině.
Maximum výskytu RB je v prvním a druhém roce věku života. Dvě třetiny nádorů jsou diagnostikovány u dětí mladších tří let. Po šestém roce života je výskyt RB výjimečný. Průměrný věk dětí v době diagnózy nádoru je 18 měsíců.
65% dětí s RB má formu unilaterální, zbývajících 35% bilaterální.
Klinické formy onemocnění a příznaky nemoci
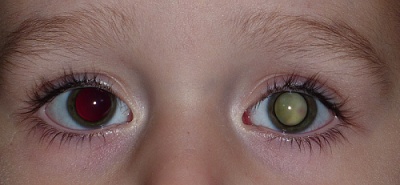
Retinoblastom postihuje jedno nebo obě oči. Všechny děti s bilaterálním postižením mají hereditární formu onemocnění. Hereditární formu onemocnění má ale i 10% dětí s unilaterálním postižením, zbývajících 90% dětí s postižením jednoho oka má formu sporadickou.
Jen 6% pacientů s hereditární formou RB má pozitivní rodinnou anamnézu s předchozím zaznamenaným výskytem retinoblastomu u blízkého příbuzného. Penetrance Rb1 genu u dětí s pozitivní rodinnou anamnézou je 80%.
Děti s hereditární formou onemocnění mívají typicky časnější výskyt onemocnění (již v prvním roce postnatálního věku) a multifokální postižení sítnice.
Průměrný věk dětí v době diagnózy u unilaterálního postižení je 2 roky, u bilaterálního postižení pak 13 měsíců.
První příznaky retinoblastomu jsou zpravidla nenápadné, v počátku probíhá onemocnění skrytě. Nádor začíná jako šedobělavé vaskularizované prominující ložisko na sítnici nebo neostrý okrsek retinálního zašednutí. V této inicální podobě je zjistitelný jen náhodně při oftalmoskopii, např. při vyšetřování sourozenců nemocného dítěte. Nejčastějším prvotním příznakem je tzv. leukokorie (šedobělavý relfex ze zornice), kdy nádor v nitru oka bez subjektivních obtíží roste dopředu a odráží světelné paprsky. Endofytický růst je charakterizován bělavými hmotami ve sklivcovém prostoru. Při exofytickém růstu působí nádor úplné odchlípení sítnice šedavé barvy s dilatovanými cévami na povrchu. Leukokorie je nejčastějším iniciálním symptomem (v 60 %) a spolu se strabismem z poruchy vidění (ve 20%) se takto manifestuje celkem až 80 % případů retinoblastomu. Každé dítě se šilháním nebo náhlým poklesem vidění musí být zavčas vyšetřeno očním lékařem k vyloučení postižení oka tímto nádorem.
Příznaky retinoblastomu vedoucí k diagnóze
| 1. | Leukokorie | 60 % | 6. | Pseudoorbitocelulitida | 3 % |
| 2. | Strabismus | 20 % | 7. | Jednostranná mydriáza | 2 % |
| 3. | Obraz nitroočního zánětu | 4 % | 8. | Heterochromie duhovky | 1 % |
| 4. | Pokles nebo ztráta vidění | 5 % | 9. | Spontánní hyféma | 1 % |
| 5. | Rutinní vyšetření | 3 % | 10. | Ostatní | 2 % |
V dalším průběhu nádor zcela vyplní nitro oka, vzniká akutní bolestivý sekundární glaukom a oko se chorobně zvětšuje. Následuje šíření nádoru přes cévnatku a bělimu, cestou zrakového nervu nebo hematogenní diseminace. Uvolňováním nádorových hmot do subretinálního prostoru se tvoří subretinální implantační metastázy prorůstající do cévnatky a extrabulbárně. Nádor má tendenci šířit se do optického nervu a do subarachnoidálního prostoru s velmi závažnou nitrolební diseminací.
Příznakem pokročilého onemocnění jsou proptóza a viditelná orbitální masa. Klinickým projevem infiltrace CNS jsou zpravidla příznaky intrakraniální hypertenze.
Stanovení diagnózy
Retinoblastom je jedním z mála dětských nádorů, u nichž není pro stanovení diagnózy nezbytné histopatologické vyšetření. Pro stanovení diagnózy retinoblastomu je postačující a zásadní vyšetření sítnice obou očí v celém rozsahu a nález nitroočního nádoru typického vzhledu.
Základem je binokulární indirektní oftalmoskopie (v mydriáze) spolu s tonometrií a biomikroskopií oka. Toto vyšetření zpravidla nelze provést bez celkové anestezie (vzhledem k věku a omezené spolupráci nemocných). Vysoký přínos má specializovaná oční sonografie, výpočetní tomografie (přesně zjistí typické kalcifikace nádoru i rozsah procesu) a MR (odhalí detailněji nitrooční nebo orbitální změny, určí podíl nádorové či tukové substance nitroočního procesu). Informativní je laboratorní vyšetření izoenzymů laktát-dehydrogenázy (LDH) v oční komorové vodě. Fluorescenční angiografie stanoví stupeň vaskularizace a aktivní kumulaci barviva v nádoru. Všechna uvedená vyšetření jsou nezbytná pro správné určení diagnózy.
Pro zjištění extraokulárního postižení je nezbytná magnetická rezonance orbit a mozku. V případě podezření na extraorbitální postižení (prorůstání tumoru do optického nervu nebo očních obalů nebo při postižení orbity) je nutné vyšetření mozkomíšního moku, spádových lymfatických uzlin, kostí a kostní dřeně k vyloučení nebo potvrzení metastatické choroby.
Diferenciální diagnóza
Diferenciálně diagnosticky je třeba odlišit další intraokulární choroby, u nichž také nacházíme leukokorii:
- perzistující primární hyperplastický sklivec,
- larvální nitrooční toxokaróza,
- Coatsova choroba (exsudativní retinální teleangiektazie),
- retrolentální fibroplazie nedonošených,
- meduloepiteliom (diktyom) řasnatého tělesa.
- retinální astrocytom, hamartom, atypický choroidální a sítnicový hemangiom
U extraokulárního onemocnění je v diferenciální diagnostice nezbytné odlišit orbitální rhabdomyosarkom, metastatický neuroblastom nebo leukemická itraorbitální infitráty.
Terapie
Léčbu retinoblastomu můžeme rozdělit na dvě základní větve – léčbu intraokulární choroby a léčbu extraokulárního postižení. Volba vhodného léčebného postupu zohledňuje i formu a rozsah onemocnění. Cílem léčby je nejen zachránit život pacienta, ale také zachránit visus a minimalizovat vznik závažných pozdních následků léčby.
Možnosti léčebného protokolu zahrnují enukleaci, chemoterapii, brachyterapii, laserovou fotokoagulaci, kryoterapii, termochemoterapii. Vždy je snaha o zachování očního bulbu s užitečným viděním, avšak asi polovina případů retinoblastomu stále vyžaduje enukleaci oka pro záchranu života dítěte.
Chirurgická léčba
Enukleace s resekcí optického nervu je nejrychlejší a nejbezpečnější léčbou retinoblastomu. Je indikována u velkého rozsahu intraokulárního postižení spojeného s nulovou šancí na záchranu zraku; a dále v případě glaukomu a invaze do přední komory. U extraorbitálního postižení může být vyjímečně indikována i exenterace orbity. V případě enukleace oka je po zhojení zaveden oční implantát s oční protézkou, která minimalizuje kosmetický defekt a napomáhá symetrickému růstu obou orbit.
Zevní radioterapie
Retinoblastom patří mezi vysoce radiosenzitivní nádory a zevní radioterapie (external beam radiotherapy - EBRT)je nejefektivnější lokální léčbou retinoblastomu. její užití je však limitována závažnými vedlejšími efekty léčby, zejména rizikem vzniku sekundárních malignit a inhibicí růstu orbity s asymetrií obličeje. Standardní dávkou EBRT je 40-45 Gy standardní frakcionací, s použitím lineárního urychlovače. Dnes je EBRT indikována vyjímečně v případě masivního postižení orbity či optického nervu.
Intraokulární terapie je zásadní léčebnou modalitou v léčbě menších nitroočních nádorů (<5mm). Zkušený oftalmolog může použít kryoterapii (zejména u tumorů v předním segmentu do velikosti 5mm a prominence do 3mm), laserovou terapii (neboli fotokoagulaci u tumorů do velikosti 5mm a prominence 3mm v zadním segmentu) nebo aplikaci radioaktivních episklerálních plaků (tzv. brachyterapii pomocí 60Co, 106Ru nebo 125I), která je účinná u nádorů menších než 15 mm při bázi, s prominencí do 10 mm. Jednotlivé přístupy mohou být navzájem kombinovány a bývají zpravidla užity opakovaně. Je možné použít i kombinace termoterapie diodovým laserem v kombinaci se systémovou chemoterapií karboplatinou (tzv. termochemoterapie se synergistickým efektem).
Chemoterapie
V současné době se chemoterapie používá v léčbě intraokulárních rozsáhlejších nádorů s cílem zmenšit jejich velikost a umožnit následnou lokální terapií záchranu oka (tzv. chemoredukce). Chemoredukce vede také ke snížení nutnosti enukleace oka a/nebo užití zevní radioterapie. Většina chemoterapeutických režimů obsahuje karboplatinu, která velmi dobře proniká dovnitř oka. Nejčastěji se používá v kombinace s Vincristinem a Etoposidem. U rozsáhlých intraokulárních nádorů po enukleaci je nutné histopatologické vyšetření enukleovaného bulbu. Retinoblastom tvoří neuroektodermální buňky s převahou jader s typickým nálezem tzv. Flexner-Wintersteinerových rozet. V případě prorůstání nádoru do optického nervu (za lamina cribrosa) nebo prorůstání do očních obalů (masivní postižení chorioidey) je nutná adjuvantní chemoterapie.
Kombinovaná systémovou chemoterapie spolu s enukleací a následnou zevní radioterapií je indikována v případě extraokulárních nádorů s intraorbitálním postižením.
U metastatických onemocnění je zpravidla indikována vysoce dávkovaná chemoterapie s autologní transplantací krvetvorné tkáně.
Sledování po léčbě
U dětí s bilaterální formou retinoblastomu se mohou nové nádory (i po adekvátní léčbě) objevovat až do 7 let věku. Do té doby musí být všechny děti po léčbě pravidelně sledovány s nezbytnými očními vyšetřeními v celkové anestezii, intervaly kontrol jsou v prvních dvou letech 14-28 dní, poté se mohou prodloužit. I děti s unilaterální formou onemocnění musí být pravidelně sledovány dle individualizovaného plánu sledování v závislosti na rozsahu onemocnění a způsobu léčby.
V případě hereditární formy nemoci je nutné celoživotní sledování a poradenství nemocnému a jeho rodině s ohledem na možný přenos onemocnění na potomstvo a riziko vzniku sekundárních malignit.
Prognóza
Prognóza onemocnění stran záchrany života je dnes v rozvinutých zemích excelentní s přežitím (EFS) 95-98 %; retinoblastom tak patří k nejúspěšněji léčitelným maligním nádorům dětského věku. Předpokladem je záchyt onemocnění v časném – intrabulbárním – stadiu. Naproti tomu pozdní záchyt onemocnění s metastatickým postižením je spojen s podstatně horší prognózou (0-40%).
Melanom
Uveální melanom je nejčastějším a nejzávažnějším zhoubným nitroočním nádorem u dospělých pacientů. Vzniká z melanocytárních buněk duhovky, řasnatého tělesa nebo cévnatky. Melanom cévnatky je nejčastější (až 80 % všech uveálních melanomů). Melanomy duhovky a řasnatého tělesa jsou méně časté (20%).
Nejčastěji se vyskytuje ve věku 50- 60 let. Vzácně byl prokázán u pacientů mladších 20 let.
Muži jsou postiženi častěji než ženy. Výskyt v evropské populaci je 2,5 / 10 tis. osob.
Rizikovými faktory jsou bílá rasa, kožní melanocytóza, neurofibromatóza, pigmentové névy, intenzívní expozice slunečnímu záření.
Melanom cévnatky nemá zpočátku choroby žádné příznaky pokud začíná růst z oblasti mimo žlutou skvrnu (místo nejostřejšího vidění, makula) a jeho zjištění bývá náhodné. S postupným růstem nádoru dochází k poruchám vidění – metamorfopsiím, snížení zrakové ostrosti a výpadům zorného pole, někdy příznaky odchlípení sítnice – blesky a jiskření. Pokud způsobí svým růstem zvýšení nitroočního tlaku (sekundární glaukom) přistupují výrazné bolesti oka. Diagnóza se stanovuje na základě podrobného oftalmoskopického a biomikroskopického vyšetření očního pozadí. Důležitá je fotodokumentace nádoru, pomocí které hodnotíme při pravidelných kontrolách změny velikosti nádoru.
Vždy je nutné pečlivě vyšetřit obě oči. Diagnostika je doplněna ultrazvukovým vyšetřením, angiografií, počítačovou tomografií a magnetickou rezonancí. Jako screeningové vyšetření k vyloučení metastáz primárního očního nádoru nebo k vyloučení metastattického karcinomu se provádí vyšetření jaterních testů, ALP, LDH, krevního obrazu, moči a sedimentu, RTG srdce a plic, ultrazvukové vyšetření břicha.
CT a NMR je nezbytné při extrabulbární expanzi melanomu. Na očním pozadí jsou počínající melanomy kopulovitého tvaru , později hřibovitého. Vzácně může být diagnostikován typ difúzního melanomu cévnatky. Pigmentace melanomů může být různě vyjádřena od sytě pigmentovaných až po tzv amelanotické. Často jsou přítomny degenerativní cystoidní změny sítnice v okolí nádoru, krvácení do sítnice nebo její totální odchlípení. Dalšími průvodními komplikacemi mohou být prokrvácení sklivce (hemoftalmus), šedý zákal (katarakta) nebo sekundární glaukom. Nádor se může šířit do sítnice a sklivce, zrakového nervu nebo prorůstat stěnou bělimy do očnice. Melanom uvey nejčastěji matastazuje do jater, plic, CNS a kostí.
Většina melanomů zůstává stacionární bez tendence metastazovat. Při prokázaném růstu se provádí brachyterapie - lokální ozáření nebo chirurgická excize nádoru. U velkých nádorů při slepém oku je nezbytná enukleace oka. Léčba je tedy závislá na celé řadě faktorů: velikost a lokalizace melanomu, stavu vidění postiženého a druhého oka, event. extrabulbární růst nádoru a přítomnost metastáz.
V léčbě se používá radioterapie-brachyterapie, Leksellův gama nůž, laserová fotokoagulace, fotodynamická terapie, chirurgická resekce. Pokud melanom prorůstá bělimou je nutná po enukleaci oka další terapie následným ozářením nebo někdy exenterace očnice.
Prognóza dalšího života závisí na histologickém typu melanomu, velikosti, lokalizaci nádoru a věku pacienta. Platí, že čím větší nádor tím horší prognóza, melanomy řasnatého tělesa mají horší prognózu než melanomy cévnatky. V případě pozdějšího vzniku metastáz u smíšeného nebo epiteloidního typu melanomu umírá do 5 let po enukleaci asi 50 % nemocných.
Maligní melanom duhovky vzniká většinou z névů duhovky. Ve většině případů bývá lokalizován v dolní polovině duhovky jako prominující různě pigmentovaný nádor. Může být ohraničený nebo difúzní a pro svou viditelnost bývá relativně včas diagnostikován. Pokud je nádor lokalizován při okraji zornice vede k jejímu zneokrouhlení. Terapií je radikální chirurgické odstranění ve zdravé tkáni duhovky.
Metastázy nádorů do cévnatky jsou relativně časté – u žen metastazuje do cévnatky karcinom prsu, u mužů pak bronchogenní karcinom. Méně časté jsou metastázy z nádorů jater, štítné žlázy, ledvin a prostaty. Tyto metastázy mohou postihovat obě oči, je tedy nezbytné pečlivé vyšetření celého rozsahu očního pozadí v mydriáze. Metastázy jsou patrné na očním pozadí je žlutobělavé uzly s okolním odchlípením sítnice. Terapie závisí na velikosti metastáz a na celkovém stavu nemocného (pravidlené sledování, chirurgické resekce, ozařování, chemoterapie).
Zhoubné nádory očnice, víček a slzné žlázy
Maligní lymfom
Maligní lymfom patří mezi nejčastější nádory očnice po 40. roce věku. Klinický obraz je charakterizován pomalu se rozvíjející protruzí oka, společně s hmatnými tuhými uzly v horních kvadrantech při orbitálním vchodu a poruchou hybnosti oka. Často bývá nádor lokalizován i v podkoží horního víčka a pod bulbární spojivkou s patrnými narůžovělými hmotami. V retrobulbárním prostoru může lymfom vytvářet ohraničená oblá ložiska nebo se může šířit difuzně kolem očního bulbu. Nádor je tvořen z monoklonálních B-lymfocytů. V polovině případů je prvním projevem celkového onemocnění. U hmatných dostupných nádorů je třeba provést biopsii pro histologické a imunologické vyšetření.
V léčbě se využívá radioterapie při vyloučení lymfomu v jiných lokalizacích. Většinou se však používá kombinovaná léčba - radioterapie a chemoterapie. Pacienti jsou v trvalé péči hematoonkologické a oční lékař pouze kontroluje lokální nález. Prognóza orbitální formy maligního lymfomu je velmi příznivá.
Rabdomyosarkom
Rabdomyosarkom je nejčastějším primárním maligním nádorem očnice v dětské populaci. Více než 70% nádorů je zjištěno v první dekádě života s vysokou frekvencí výskytu v prvním roce života. Nádor se klinicky manifestuje jako rychle rostoucí protruze často s dislokací bulbu, pseudozánětlivým příznaky (edém a zarudnutí víček, chemóza bulbární spojivky, bolest) a výraznou poruchou hybnosti. Tento klinický obraz se podobá orbitocelulitidě. Nejsou však žádné celkové symptomy a počítačová tomografie vyloučí paranazální sinusitidu.
Na tomogramu jsou pro rabdomyosarkom typické známky destrukce přilelhlé části skeletu očnice. Vzácněji najdeme, že nádor prorůstá do paranazálních dutin a extraorbitálně skrze obě fisury. Magnetická rezonance a počítová tomografie s kontrastní látkou prokazuje masu tumoru s intenzivním „enhancement“ Při podezření na rabdomyosarkom je nutné zavčas provést biopsii se zafixováním vzorku tumoru ve formaldehydu nebo glutaraldehydu na histologické a elektronmikroskopické vyšetření a verifikaci klinické diagnózy. Patologicko-anatomicky rozeznáváme tyto typy rabdomyosarkomu: Embryonální typ (65% případů) je nejčastější, vyskytuje nejvíce u nejmenších dětí, jeho predilekční lokalizací v očnici je nazální horní kvadrant za orbitálních vchodem nebo retrobulární prostor. Působí protruzi s dislokací temporálně dolů a pseudptózu. Při včasném záchytu a včas zahájené komplexní léčbě má tento typ relativně dobrou životní prognózu. Alveolární typ (30% případů) se vyskytuje spíše u starších dětí, je vzácnější, prognosticky daleko závažnější než embryonální typ. Typickou lokalizací je spodina očnice, působí protruzi s dislokací bulbu nahoru. Pleomorfní typ (5%) se vyskytuje u dětí extrémně vzácně. Je častější u dospělých a jeho prognóza je většinou infaustní.
Diferernciálně diagnosticky nutno odlišit orbitocelulitidu, metastázy neuroblastomu, angioblastický hemangiom, orbitální projevy akutní lymfatické leukémie, krvácení do lymfangiomu, ale i vzácný maligní teratom.
Kromě histologického typu rabdomyosarkomu je pro zhoršení životní prognózy významná expanze tumoru do paranazálních dutin a jeho parameningální šíření do intrakraniálního prostoru. Metastázy krevní cestou do plic jsou dosti časté a vznikají brzy, ale některé studie neprokázaly tento faktor jako signifikantní pro určování životní prognózy.
V 70.letech minulého století po chirurgické léčbě subtotální extirpací tumoru nebo exenterací očnice přežívalo pouze 20% dětských pacientů. V současné době po kombinované léčbě chirurgické s radioterapií a chemoterapií je 5-leté přežití prokázáno u 94% ze souboru 221 dětských pacientů s embryonálním rabdomyosarkomem. V případě alveolárního typu klesá tento počet na 74% Kombinovaná léčba (chirurgie- chemoterapie- radioterapie) je aktuálně modifikována podle mezinárodních léčebných protokolů (Intergroup Rhabdomyosarkoma Study, IRS). Pro jednotlivé histologické typy jsou vypracována speficifická léčebná schemata kombinací chemoterapeutik. Léčba rabdomyoskarkomu dětské očnice je vždy vedena týmem v němž nemůže chybět onkolog, pediatr, oftalmolog, radiolog a histolog. Recidiva orbitálního nádoru je závažným terapeutickým problémem. V současné době se doporučuje v tomto případě široká excize postižené tkáně s okolní kostí event. paranazální dutinou. Následuje chemoterapie a pokud nebyla provedena dříve, tak radioterapie. Prognóza dlouhodobého přežití u těchto nemocných je méně úspěšná. Sekundární malignity jako např. leukémie nejsou vzácností.
Maligní nádory víček
Bazocelulární karcinom (bazaliom) je nečastějším zhoubným nádorem víček (90 %). Histologicky rozlišujeme několik desítek typů těchto nádorů. Většinou je bazaliom lokalizován na dolním víčku při vnitřním koutku. Charakteristické je opakovaná tvorba ulcerací v centru nádoru. Bazaliomy nevytvářejí metastázy, ale infiltrativně prorůstají hlubšími vrstvami podkoží do očnice.
Spinocelulární karcinom (spinaliom) se vyskytuje vzácněji – asi 10% všech zhoubných nádorů víček.
Klinický obraz je značně variabilní – nejčastěji tuhý nádorový uzel se šupinami z povrchových vrstev kůže. Diagnózu je možno potvrdit pouze na základě histologického vyšetření. Nádor může metastázovat do regionálních lymfatických uzlin.
Základem léčby je chirurgická resekce celého tumoru bezpečně do zdravé tkáně.
Doplňující léčbou je radioterapie, kryoterapie či laserová fotokoagulace.
Maligní nádory slzné žlázy
Adenoidně cystický karcinom (cylindrom) je zastoupen 25% všech nádorů slzné žlázy. Je charakterizován vysokou malignitou, mimořádným sklonem k recidivám a vysokou mortalitou.
Pleomorfní adenokarcinom (maligní smíšený nádor) je vzácnější, méně maligní. Klinický obraz jednotlivých morfologických forem se zpočátku příliš neliší. Maligní nádory slzné žlázy jsou na pohmat i spontánně velmi bolestivé. Dislokace očního bulbu vede ke dvojitému vidění, může být překrvení, otok spojivky a esovitá deformace horního víčka.
Terapie obou zhoubných typů nádorů slzné žlázy spočívá pouze v jejich radikální chirurgické exstirpaci. Ostatní léčebné možnosti jsou v podstatě neúčinné. Biopsie nebo subtotální exstirpace je kontraindikována pro riziko rychlé diseminace nádoru, která je dále léčebně neovlivnitelná.
Prognóza dlouhodobého přežití je u karcinomů slzné žlázy velmi špatná, a to i při adekvátní chirurgické resekci včetně exenterace očnice. Důvodem je perineurální šíření tumoru a recidivy intrakraniálně. Recidivy tumoru jsou rezistentní na radioterapii a chemoterapii.