Konference: 2015 20th Congress of the European Hematology Association - účast ČR
Kategorie: Myeloproliferativní nemoci
Téma: Poster
Číslo abstraktu: P371
Autoři: Mgr. Pavla Korálková (Pospíšilová); Mgr. Renata Mojzíková (Solná), Ph.D.; MUDr. Pavel Timr; Dr. Joan Lluis Vives Corrons; Dr. Christine Macartney; Doc.RNDr. Vladimír Divoký, Ph.D.; MD Richard van Wijk
Background
Hexokinase (HK) deficiency is a very rare cause of hereditary nonspherocytic hemolytic anemia (HNSHA). Hexokinase is one of the key regulatory enzymes of glycolysis, on which the red blood cell is totally dependent the production of ATP. Deficiency of HK disrupts cellular metabolism, which ultimately results in HNSHA. To date, 24 cases of hexokinase deficiency have been described. Only four of them have been characterized on the DNA level. We here describe five new cases and 3 novel mutations in HK1.
Aims
Characterize a genetic defect in patients who were found to be HK-deficient by spectrophotometrically determined red blood cell HK and pyruvate kinase (PK) enzymatic activities.
Methods
PK/HK ratio was applied to evaluate the effect of age-related increases in enzymatic activity. To confirm the diagnosis, DNA sequence analysis of HK1 was performed by Sanger sequencing. Novel mutations were characterized by biochemical methods (Km for glucose and Mg.ATP, pH stability and thermolability) and molecular studies (RT-PCR, quantitative RT-PCR, and Western Blot analysis on ex vivo cultured patient erythroblasts).
Results
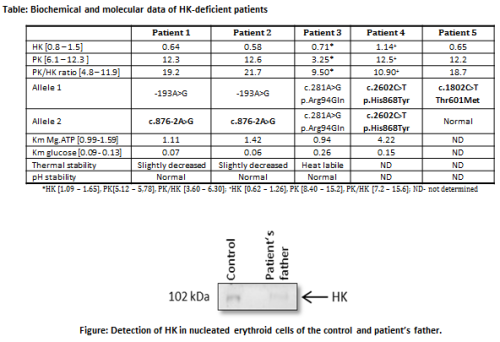
DNA sequence analysis of
HK1 of 5 patients with suspected HK deficiency revealed a
total of 5 different mutations in HK1. Three of them were
novel (Table, novel mutations in bold). In silico analysis
of mutations p.His868Tyr and p.Thr601Met by Polyphen-2 and SIFT
predict that both substitutions are not tolerated. Both are located
in the catalytic region, and kinetic properties are likely to be
impaired upon mutation. Kinetic studies of the p.His868Tyr HK
mutant showed that its affinity for ATP was indeed markedly
decreased (3.2-times), whereas the affinity for glucose was
slightly increased (Table). The molecular effects of the novel
splice site mutation c.876-2A>G in intron 7 was studied on mRNA
isolated from ex vivocultured erythroblasts from the
patient’s father, who was heterozygous for this mutation. RT-PCR
analysis showed the presence of normal as well as two aberrant
mRNAs species. The aberrantly spliced transcripts lacked
either exon 8 or both exons 8 and 9. Quantitative RT-PCR analysis
showed that expression levels of the normally spliced mRNA variant
were down-regulated 3-times compared to normal. These findings were
confirmed on the protein level by Western blot analysis of HK from
nucleated erythroid cells (Figure).
Summary
We report 5 mutations in HK1 in 4 unrelated families.
Three of them, c.876-2A>G, Thr601Met, and p.His868Tyr have not
been previously reported. The pathogenic nature of novel mutations
was confirmed by molecular and biochemical studies. Our results
contribute to a better understanding of the genotype-to-phenotype
correlation of HK deficiency, a very rare enzyme disorder of the
red blood cell. Supported by Ministry of Health of
Czech Republic, grant NT/13587 and IGA_LF_2015_015.
Keyword(s): Hemolytic anemia, Red blood cell
Datum přednesení příspěvku: 12. 6. 2015