Konference: 2015 57th ASH Annual Meeting - účast ČR
Kategorie: Maligní lymfomy a leukémie
Téma: 112. Thalassemia and Globin Gene Regulation: Poster III
Číslo abstraktu: 3371
Autoři: N. Scott Reading, Ph.D.; Dr. Barnaby E. Clark, Ph.D.; Jihyun Song, MS; Claire C Shooter; MD Robin E. Miller; Dr. Archana M. Agarwal, MBBS; MD Swee Lay Thein; Doc.RNDr. Vladimír Divoký, Ph.D.; MD Josef T. Prchal
We report a unique head-to-tail duplication of the β-globin cluster in a patient phenotypically expressing homozygous HbS (sickle-cell anemia, SCA) that provides insight into the regulatory role of the β-LCR and 3’HS1 on wild-type β-globin (β-A) expression in a background of SCA. The studies were driven by an apparent discrepancy between hemoglobin analysis of an infant with a SCA phenotype and no detectable HBA, and Sanger sequencing of the β-globin genes which showed a heterozygous genotype.
Analyses of parents’ blood samples and DNA revealed that each were carriers for sickle cell allele. Hemoglobin analysis showed the father expressed HbS fraction at 41.3% and the mother at 33.3%, while the proband had a majority of HbS, some HbF and no detectable HbA. The reduced HbS fraction in the mother could be explained by co-inheritance of α-thalassemia (αα/α-3.7). The proband did not inherit α-thalassemia from the mother.
Multiplex ligation-dependent probe amplification analysis of the proband’s DNA suggested duplication of the β-globin cluster, resulting in three copies of the HBB gene in the genome. Subsequent next-generation sequencing confirmed that the duplication occurred immediately adjacent to the first iteration of sequence, in head-to-tail orientation and resulted in an intact β-S cluster having both LCR and HS1 elements, followed by the duplicated β-A cluster (β-S, β-A) that excluded a part of HBE (epsilon globin) and the upstream β-LCR regions, extending through to LINE L1LBP1 ([hg19] chr11:4640332–5290168). Further analyses revealed that the duplicated β-A cluster, which encompassed approximately 650 kb sequence, lacked a DNA segment containing the 3’HS1 element (figure 1). The proband’s β genotype is thus (β-S/β-S, β-A). DNA analysis showed that the father carried the duplicated β-globin cluster with genotype (β-A/β-S, β-A), and the mother, a heterozygous HbS genotype (β-A/β-S).
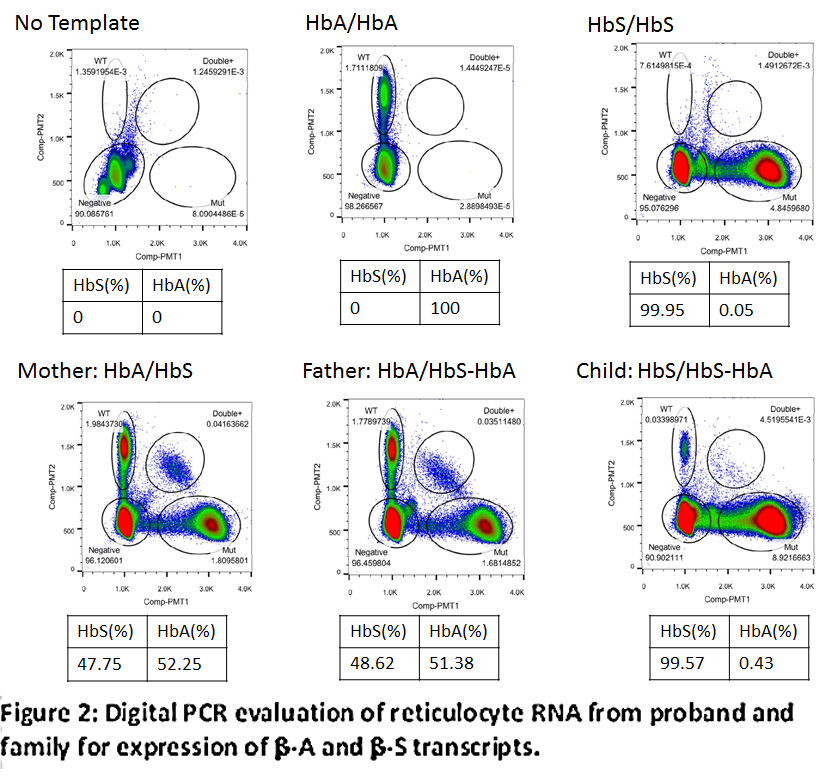
Reverse transcription, quantitative polymerase chain reaction (RT-qPCR) was used to assess transcription levels of β-A and β-S mRNA for each family member. Analysis of the parents’ reticulocyte RNA showed that the β-globin (β-A and β-S) transcript levels were nearly balanced. RT-qPCR of proband’s reticulocyte RNA showed no convincing detection of β-A transcript, but the β-A transcript was clearly detected by digital PCR, albeit at a very low level (0.4% of total HBB transcript) (figure 2).
The β-globin cluster duplication on one chromosome in a background of a phenotypically homozygous (HbSS) SCA patient has provided a unique opportunity to assess the effect of the LCR and 3’HS1 regions on the transcription of a β-A gene in an unbiased environment. Prior studies in transformed cell-lines or mouse models have shown the down regulatory effect of LCR loss and potential protective effect of the 3’HS1 element. Within the human model, the observed transcription from the duplicated, distally displaced (~650 kb) β-A cluster demonstrates that the loss of LCR and flanking HS sites does not lead to complete silencing of β-globin transcription.
VD was supported by project IGA MzCR NT13587.

Datum přednesení příspěvku: 7. 12. 2015