Konference: 2005 XXIX. Brněnské onkologické dny a XIX. Konference pro sestry a laboranty
Kategorie: Nádorová biologie/imunologie/genetika a buněčná terapie
Téma: Pokroky v molekulární biologii nádorů
Číslo abstraktu: 056
Autoři: Mgr. Jitka Petrlová, Ph.D.; O. Blaštík; doc.RNDr. Vojtěch Adam, Ph.D.; R. Mikelová; Mgr. David Potěšil; MUDr. Lucia Trnková; Prof. MUDr. Richard Průša, CSc.; doc.Ing. René Kizek, Ph.D.
Klíčová slova
Metalothionein; kovy vázající proteiny, visící kapková rtuťová elektroda, chronopotenciometrická rozpouštěcí analýza, katalytický signál, pík H, prenátriová vlna; krevní sérum, lidská moč
1. Úvod
Objev metalothioneinu (MT) je datován rokem 1957, kdy Margoshes a Valee izolovali MT z koňských ledvin. Metalothionein patří do skupiny intracelulárních, nízkomolekulárních na cystein velmi bohatých proteinů (obsah Cys až 30 % v molekule proteinu) o molekulové hmotnosti 6-10 kDa. V molekule MT nejsou přítomny aromatické aminokyseliny a cysteiny se v primární sekvenci vyskytují obvykle v těchto repeticích: Cys-X-Cys, Cys-Cys-X-Cys-Cys, Cys-X-Cys-Cys, kde X představuje jinou aminokyselinu než cystein. MT se skládají ze dvou vazebných domén α,β, které jsou složeny z cysteinových klastrů. N-terminální část peptidu je označena jako β-doména, má tři vazebná místa pro dvojmocné ionty; C-terminální část peptidu (α-doména) má schopnost vyvázat čtyři dvojmocné ionty kovů. V případě jednomocných iontů kovů je MT schopen vázat celkem 12 atomů. Hlavní biologickou funkcí MT, díky své vysoké afinitě k těžkým kovům, např. k zinku, mědi nebo kadmiu, je homeostatická kontrola a detoxikace těchto těžkých kovů u vývojově rozdílných organismů. Často diskutovanou otázkou je schopnost MT transportovat atomy kovů k apoenzymům a jejich aktivní zapojení do homeostázy esenciálních prvků. Je známo, že těžké kovy produkují reaktivní kyslíkové radikály. Tyto vzniklé radikály jsou pravděpodobně schopny reagovat s MT. Význam detoxikace těžkých kovů narůstá, protož e vě tšina z tě chto těž kých kovů se rozptyluje v atmosféře nebo rozpouští ve vodách a postupně kontaminuje ekosystémy. Stálé zvyšování koncentrace těžkých kovů v životním prostředí je vážným hygienickým problémem, a proto se hledají nové metody pro jejich snadnou a rychlou detekci, použitelné i mimo specializované laboratoře.
Pro tyto účely byla využita řada analytických metod, jako je atomová absorpční spektrofotometrie, atomová absorpční spektrometrie s indukčně vázaným plazmatem, ale i elektrochemie. Cílem této práce bylo vysoce senzitivní stanovení metalothioneinu v biologické matrici, jako je lidská moč a krevní sérum.
2. Materiál a Metody
Chemikálie
MT z králičích jater (MW 7143), obsahující 5,9 % Cd a 0,5 % Zn, byl zakoupen u firmy Sigma Aldrich (St. Louis, USA). Tris(2-carboxyethyl)phosphin (TCEP) byl vyroben Molecular Probes (Evgen, Oregon, USA). Chlorid sodný a ostatní použité chemikálie byly získány v čistotě ACS od společnosti Sigma Aldrich. Zásobní roztok standardu MT 10 µg.ml–1 byl připraven za využití ACS vody (Sigma-Aldrich, USA) a uložen ve tmě při -20°C. Pracovní standardní roztoky byly vždy připravovány denně a to naředěním ze zásobního roztoku.
Vzorek lidského krevního séra byl získán z oddělení klinické biochemie, Úrazová nemocnice Ponávka v Brně, Česká republika. Vzorky byly denaturovány při teplotě 99 °C v termomixeru (Eppendorf 5430, USA) po 15 min a mícháním 350 rpm, a následně ochlazen na 4 °C. Denaturované homogenáty byly centrifugovány při 4 °C, 15 000 g po dobu 30 min. (Eppendorf 5402, USA). Tepelnou úpravou byly velmi efektivně odstraněny všechny interferující proteiny. Vzorky lidské moče byly získány od zdravých jedinců (personálu laboratoře). Získaná moč byla filtrována přes teflonový diskový filtr (0,45 µm a 13 mm průměr, Alltech Associates, Deerfield, Il, USA). Pro výpočet osmolality bylo využito faktu, že osmolalita lidské moči je 250 mmol.kg–1.
Elektrochemické stanovení bylo provedeno pomocí přístroje AUTOLAB analyzátor (EcoChemie, Netherlands), který byl napojený na VA-Stand 663 (Metrohm, Switzerland), používala se pracovní cela s tříelektrodovým systémem. Pracovní elektrodou byla visící rtuťová kapková elektroda (HMDE) s plochou kapky 0,4 mm2. Referentní elektroda byla Ag/AgCl/3M KCl a pomocná elektroda byla uhlíková tyčinka. Základní elektrolyt byl připraven smícháním jednotlivých složek pufru. Před vlastní elektrochemickou analýzou byl z roztoku odstraněn kyslík probubláním argonem po dobu 240 s. Teplota základního elektrolytu byla kontrolována pomocí termostatované nádobky za využití termostatu Ultra-thermostat U10.
3. Výsledky a diskuze
Stanovením MT pomocí CPSA v kombinaci s AdTS jsme se zabývali v našich předešlých pracích, kde jsme zjistili, že na probíhajícím katalytickém procesu se jednak účastní složky základního elektrolytu, ale také je výrazně ovlivněn protonizací MT.
Vliv redukčního činidla
K 500 fmol množství MT byl přidán 1 mM TCEP. Pozorovaný signál MT se zvýšil o více jak o 50 %. Navíc nás zajímalo, zda námi vybraná koncentrace TCEP je nejvhodnější pro stanovení MT. Proto jsme postupně přidávali rozdílnou koncentraci TCEP (0,1–1,5 mM) k MT. Pozorovaný signál MT se postupně s rostoucí koncentrací TCEP posouval směrem k pozitivním potenciálům a zvyšoval se do koncentrace TCEP 1 mM. Po dosažení tohoto maxima se pík H měnil již minimálně. Pozorovaný nárůst pravděpodobně souvisí s úplnou redukcí oxidovaných SH skupin v MT.
Analýza MT pomocí přenosové techniky
Použitím rtuťové elektrody a píku H jsme získali 3-4 krát vyšší citlivost jak u ostatních elektrochemických techniky. Pokud jsme analyzovali rozdílné velmi nízké koncentrace redukovaného MT (od 100 nM) u doby akumulace 120 s a teplotně kontrolovaném roztoku základního elektrolytu (25 °C), tak jsme získali CPSA typickou koncentrační závislost, jak je ukázáno na Obr. 1. Závislost měla tvar Langmuierovské izotermy. Tuto pozorovanou koncentrační závislost bylo možné rozdělit na dva samostatné lineární úseky (0,6 – 10 fmol v 5 µl kapce) y = 1954,5x + 38600; R2 = 0,9926 (není ukázáno). Na vloženém obrázku 1 je ukázána závislost 10 – 320 amol v 5 µl kapce (y = 54,884x + 18567; R2 = 0,9937). I při těchto velmi nízkých množstvích MT (10, 150 a 320 amol) byly pozorované píky velmi úzké, dobře vyvinuté, separované a vlastní analýza proběhla do 5 min (vložený obrázek 1). Pozorovaná relativní směrodatná odchylka se pohybovala kolem 6,5 % (n=10). Díky tomuto našemu nově modifikovanému postupu bylo možné určit limit detekce 3S/N metalothioneinu v čistém základním elektrolytu jako 11 zmol.
Analýza metalothioneinu v biologické matrici
Námi popsanou velmi senzitivní metodiku AdTS CPSA pro stanovení MT jsme dále využili ke stanovení MT ve složité biologické matrici (jako model nám posloužila lidská moč a krevní sérum). Nejdříve jsme se pokusili stanovit MT v moči. Zjistili jsme, že s narůstající osmolalitou moči dochází k poklesu signálu MT a pík MT se také posouvá k negativnějším potenciálům.
(Obr.2).
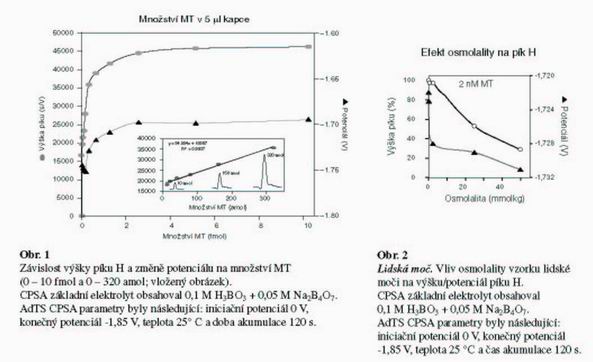
Při osmolalitě moči nad 50 mmol/kg již není vhodné provádět stanovení (pozorovaný signál se snížil o více jak 50 %). Proto je pro analytické stanovení proteinů (MT) pomocí AdTS CPSA potřebné studovaný biologický vzorek vhodně naředit, aby byla snížena jeho specifická hustota a senzitivita stanovení zůstala zachována. K moči (0,25 mmol/kg) bylo postupně přidáváno rozdílné množství MT. Získali jsme lineární závislost na koncentraci (y = 11317x + 9133,4; R2 = 0,9994).
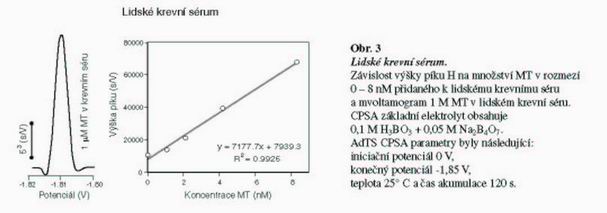
Nedávno bylo zjištěno, že hladina MT se mění u závažných onemocnění jater, jako je hepatocelulární karcinom, chronická hepatitida a cirhóza jater. Hladina MT byla stanovena pomocí ELISA po kyselé a tepelné úpravě vzorku. My jsme se rozhodli využít naší AdTS CPSA techniky pro stanovení MT v lidském krevním séru. Nejdříve byl vzorek tepelně denaturován a po centrifugaci byl odebraný supernatant použitý pro analýzu MT. Vzorek byl před vlastní analýzou naředěn 1000x a následně manalyzován v 5 µl kapce. V případě analýzy více koncentrovaného vzorku dochází k poklesu píku o 25 % (v porovnáním s 10x ředěným vzorkem), pravděpodobně v důsledku adsorpce na povrch HMDE. Do takto připraveného vzorku krevního séra jsme přidávali rozdílnou koncentraci MT (Obr. 3). Získali jsme velmi dobrou lineární závislost standardního přídavku na zvyšující se koncentraci MT (y = 7177,7x + 7939,3; R2 = 0,9925). Limit detekce se pohyboval kolem 350 fmol. V námi testovaných vzorcích lidského séra byla zjištěna průměrná koncentrace MT 1,1 ± 0,015 µM (n=5). Na obrázku Obr. 3 je ukázán chronopotenciogram odpovídající 1 µM MT koncentraci.
Lidské krevní sérum
4. Závěr
Jak se ukazuje katalytické signály vylučování vodíku umožňují také velmi senzitivní analýzu MT v atomolární koncentraci. Studovat množství MT v biologických vzorcích je velmi potřebné a jak se v posledních letech ukazuje, je hladina MT nejen markerem vlivu těžkých kovů na organismu, ale také markerem agresivních nádorových onemocnění. Námi popsaná elektroanalytická metoda AdTS CPSA umožňuje stanovení MT v zeptomolech v borátovém pufru, pH 8,0 a je jí možné aplikovat na analýzu reálných vzorků, jako je lidské krevní sérum a moč.
Literatura
Příspěvek vznikl za podpory grantů RASO 2005 a GAČR 525/04/P132
Metalothionein; kovy vázající proteiny, visící kapková rtuťová elektroda, chronopotenciometrická rozpouštěcí analýza, katalytický signál, pík H, prenátriová vlna; krevní sérum, lidská moč
1. Úvod
Objev metalothioneinu (MT) je datován rokem 1957, kdy Margoshes a Valee izolovali MT z koňských ledvin. Metalothionein patří do skupiny intracelulárních, nízkomolekulárních na cystein velmi bohatých proteinů (obsah Cys až 30 % v molekule proteinu) o molekulové hmotnosti 6-10 kDa. V molekule MT nejsou přítomny aromatické aminokyseliny a cysteiny se v primární sekvenci vyskytují obvykle v těchto repeticích: Cys-X-Cys, Cys-Cys-X-Cys-Cys, Cys-X-Cys-Cys, kde X představuje jinou aminokyselinu než cystein. MT se skládají ze dvou vazebných domén α,β, které jsou složeny z cysteinových klastrů. N-terminální část peptidu je označena jako β-doména, má tři vazebná místa pro dvojmocné ionty; C-terminální část peptidu (α-doména) má schopnost vyvázat čtyři dvojmocné ionty kovů. V případě jednomocných iontů kovů je MT schopen vázat celkem 12 atomů. Hlavní biologickou funkcí MT, díky své vysoké afinitě k těžkým kovům, např. k zinku, mědi nebo kadmiu, je homeostatická kontrola a detoxikace těchto těžkých kovů u vývojově rozdílných organismů. Často diskutovanou otázkou je schopnost MT transportovat atomy kovů k apoenzymům a jejich aktivní zapojení do homeostázy esenciálních prvků. Je známo, že těžké kovy produkují reaktivní kyslíkové radikály. Tyto vzniklé radikály jsou pravděpodobně schopny reagovat s MT. Význam detoxikace těžkých kovů narůstá, protož e vě tšina z tě chto těž kých kovů se rozptyluje v atmosféře nebo rozpouští ve vodách a postupně kontaminuje ekosystémy. Stálé zvyšování koncentrace těžkých kovů v životním prostředí je vážným hygienickým problémem, a proto se hledají nové metody pro jejich snadnou a rychlou detekci, použitelné i mimo specializované laboratoře.
Pro tyto účely byla využita řada analytických metod, jako je atomová absorpční spektrofotometrie, atomová absorpční spektrometrie s indukčně vázaným plazmatem, ale i elektrochemie. Cílem této práce bylo vysoce senzitivní stanovení metalothioneinu v biologické matrici, jako je lidská moč a krevní sérum.
2. Materiál a Metody
Chemikálie
MT z králičích jater (MW 7143), obsahující 5,9 % Cd a 0,5 % Zn, byl zakoupen u firmy Sigma Aldrich (St. Louis, USA). Tris(2-carboxyethyl)phosphin (TCEP) byl vyroben Molecular Probes (Evgen, Oregon, USA). Chlorid sodný a ostatní použité chemikálie byly získány v čistotě ACS od společnosti Sigma Aldrich. Zásobní roztok standardu MT 10 µg.ml–1 byl připraven za využití ACS vody (Sigma-Aldrich, USA) a uložen ve tmě při -20°C. Pracovní standardní roztoky byly vždy připravovány denně a to naředěním ze zásobního roztoku.
Vzorek lidského krevního séra byl získán z oddělení klinické biochemie, Úrazová nemocnice Ponávka v Brně, Česká republika. Vzorky byly denaturovány při teplotě 99 °C v termomixeru (Eppendorf 5430, USA) po 15 min a mícháním 350 rpm, a následně ochlazen na 4 °C. Denaturované homogenáty byly centrifugovány při 4 °C, 15 000 g po dobu 30 min. (Eppendorf 5402, USA). Tepelnou úpravou byly velmi efektivně odstraněny všechny interferující proteiny. Vzorky lidské moče byly získány od zdravých jedinců (personálu laboratoře). Získaná moč byla filtrována přes teflonový diskový filtr (0,45 µm a 13 mm průměr, Alltech Associates, Deerfield, Il, USA). Pro výpočet osmolality bylo využito faktu, že osmolalita lidské moči je 250 mmol.kg–1.
Elektrochemické stanovení bylo provedeno pomocí přístroje AUTOLAB analyzátor (EcoChemie, Netherlands), který byl napojený na VA-Stand 663 (Metrohm, Switzerland), používala se pracovní cela s tříelektrodovým systémem. Pracovní elektrodou byla visící rtuťová kapková elektroda (HMDE) s plochou kapky 0,4 mm2. Referentní elektroda byla Ag/AgCl/3M KCl a pomocná elektroda byla uhlíková tyčinka. Základní elektrolyt byl připraven smícháním jednotlivých složek pufru. Před vlastní elektrochemickou analýzou byl z roztoku odstraněn kyslík probubláním argonem po dobu 240 s. Teplota základního elektrolytu byla kontrolována pomocí termostatované nádobky za využití termostatu Ultra-thermostat U10.
3. Výsledky a diskuze
Stanovením MT pomocí CPSA v kombinaci s AdTS jsme se zabývali v našich předešlých pracích, kde jsme zjistili, že na probíhajícím katalytickém procesu se jednak účastní složky základního elektrolytu, ale také je výrazně ovlivněn protonizací MT.
Vliv redukčního činidla
K 500 fmol množství MT byl přidán 1 mM TCEP. Pozorovaný signál MT se zvýšil o více jak o 50 %. Navíc nás zajímalo, zda námi vybraná koncentrace TCEP je nejvhodnější pro stanovení MT. Proto jsme postupně přidávali rozdílnou koncentraci TCEP (0,1–1,5 mM) k MT. Pozorovaný signál MT se postupně s rostoucí koncentrací TCEP posouval směrem k pozitivním potenciálům a zvyšoval se do koncentrace TCEP 1 mM. Po dosažení tohoto maxima se pík H měnil již minimálně. Pozorovaný nárůst pravděpodobně souvisí s úplnou redukcí oxidovaných SH skupin v MT.
Analýza MT pomocí přenosové techniky
Použitím rtuťové elektrody a píku H jsme získali 3-4 krát vyšší citlivost jak u ostatních elektrochemických techniky. Pokud jsme analyzovali rozdílné velmi nízké koncentrace redukovaného MT (od 100 nM) u doby akumulace 120 s a teplotně kontrolovaném roztoku základního elektrolytu (25 °C), tak jsme získali CPSA typickou koncentrační závislost, jak je ukázáno na Obr. 1. Závislost měla tvar Langmuierovské izotermy. Tuto pozorovanou koncentrační závislost bylo možné rozdělit na dva samostatné lineární úseky (0,6 – 10 fmol v 5 µl kapce) y = 1954,5x + 38600; R2 = 0,9926 (není ukázáno). Na vloženém obrázku 1 je ukázána závislost 10 – 320 amol v 5 µl kapce (y = 54,884x + 18567; R2 = 0,9937). I při těchto velmi nízkých množstvích MT (10, 150 a 320 amol) byly pozorované píky velmi úzké, dobře vyvinuté, separované a vlastní analýza proběhla do 5 min (vložený obrázek 1). Pozorovaná relativní směrodatná odchylka se pohybovala kolem 6,5 % (n=10). Díky tomuto našemu nově modifikovanému postupu bylo možné určit limit detekce 3S/N metalothioneinu v čistém základním elektrolytu jako 11 zmol.
Analýza metalothioneinu v biologické matrici
Námi popsanou velmi senzitivní metodiku AdTS CPSA pro stanovení MT jsme dále využili ke stanovení MT ve složité biologické matrici (jako model nám posloužila lidská moč a krevní sérum). Nejdříve jsme se pokusili stanovit MT v moči. Zjistili jsme, že s narůstající osmolalitou moči dochází k poklesu signálu MT a pík MT se také posouvá k negativnějším potenciálům.
(Obr.2).
Při osmolalitě moči nad 50 mmol/kg již není vhodné provádět stanovení (pozorovaný signál se snížil o více jak 50 %). Proto je pro analytické stanovení proteinů (MT) pomocí AdTS CPSA potřebné studovaný biologický vzorek vhodně naředit, aby byla snížena jeho specifická hustota a senzitivita stanovení zůstala zachována. K moči (0,25 mmol/kg) bylo postupně přidáváno rozdílné množství MT. Získali jsme lineární závislost na koncentraci (y = 11317x + 9133,4; R2 = 0,9994).
Nedávno bylo zjištěno, že hladina MT se mění u závažných onemocnění jater, jako je hepatocelulární karcinom, chronická hepatitida a cirhóza jater. Hladina MT byla stanovena pomocí ELISA po kyselé a tepelné úpravě vzorku. My jsme se rozhodli využít naší AdTS CPSA techniky pro stanovení MT v lidském krevním séru. Nejdříve byl vzorek tepelně denaturován a po centrifugaci byl odebraný supernatant použitý pro analýzu MT. Vzorek byl před vlastní analýzou naředěn 1000x a následně manalyzován v 5 µl kapce. V případě analýzy více koncentrovaného vzorku dochází k poklesu píku o 25 % (v porovnáním s 10x ředěným vzorkem), pravděpodobně v důsledku adsorpce na povrch HMDE. Do takto připraveného vzorku krevního séra jsme přidávali rozdílnou koncentraci MT (Obr. 3). Získali jsme velmi dobrou lineární závislost standardního přídavku na zvyšující se koncentraci MT (y = 7177,7x + 7939,3; R2 = 0,9925). Limit detekce se pohyboval kolem 350 fmol. V námi testovaných vzorcích lidského séra byla zjištěna průměrná koncentrace MT 1,1 ± 0,015 µM (n=5). Na obrázku Obr. 3 je ukázán chronopotenciogram odpovídající 1 µM MT koncentraci.
Lidské krevní sérum
4. Závěr
Jak se ukazuje katalytické signály vylučování vodíku umožňují také velmi senzitivní analýzu MT v atomolární koncentraci. Studovat množství MT v biologických vzorcích je velmi potřebné a jak se v posledních letech ukazuje, je hladina MT nejen markerem vlivu těžkých kovů na organismu, ale také markerem agresivních nádorových onemocnění. Námi popsaná elektroanalytická metoda AdTS CPSA umožňuje stanovení MT v zeptomolech v borátovém pufru, pH 8,0 a je jí možné aplikovat na analýzu reálných vzorků, jako je lidské krevní sérum a moč.
Literatura
- M. Margoshes, B.L.A. Vallee, J. Am. Chem. Soc. 79 (1957)
4813–4814.
- I. Sestakova, M. Kopanica, L. Havran, E. Palecek,
Electroanalysis 12 (2000) 100.
- M. Tomschik, L. Havran, M. Fojta, E. Paleček, Electroanalysis
10 (1998) 403–409.
- M. Tomschik, L. Havran, E. Paleček, M. Heyrovský,
Electroanalysis 12 (2000) 274–278.
- L. Trnkova, R. Kizek, J. Vacek, Bioelectrochemistry 56 (2002)
57.
- J. Vacek, J. Petrek, R. Kizek, L. Havel, B. Klejdus, L.
Trnkova, F. Jelen, Bioelectrochemistry 63 (2004)
347–351.
- M.J. Honeychurch, M.J. Ridd, Electroanalysis 8 (1996)
654.
- R. Prusa, R. Kizek, J. Vacek, L. Trnkova, J. Zehnalek, Clin.
Chem. 50 (2004) A28.
- M. Strouhal, R. Kizek, J. Vacek, L. Trnková, M. Němec,
Bioelectrochemistry 60 (2003) 29–36.
- R. Kizek, L. Trnkova, E. Palecek, Anal. Chem. 73 (2001)
4801.
- M. Brazdova, R. Kizek, L. Havran, E. Palecek,
Bioelectrochemistry 55 (2002) 115–118.
- R. Kizek, J. Vacek, V. Adam, B. Vojtesek, Klin. Biochem. Metab.
13 (2004) 72.
- E. Palecek, M. Masarik, R. Kizek, D. Kuhlmeier, J. Hassmann, J.
Schulein, Anal. Chem. 76 (2004) 5930.
- L. Havran, S. Billova, E. Palecek, Electroanalysis 16 (2004)
1139.
- A. Nakayama, H. Fukuda, M. Ebara, H. Hamasaki, K. Nakajima, H.
Sakurai, Biological and Pharmaceutical Bulletin 25 (2002)
426.
- J. Zelená, D. Potešil, J. Vacek, V. Adam, J. Hradecký, R.
Prusa, R. Kizek, B. Vojtěšek, Klinická onkologie 17 (2004)
190.
- Y.A. Zolotov, J. Anal. Chem. 59 (2004) 807.
Příspěvek vznikl za podpory grantů RASO 2005 a GAČR 525/04/P132
Datum přednesení příspěvku: 26. 5. 2005