Konference: 2012 XXXVI. Brněnské onkologické dny a XXVI. Konference pro sestry a laboranty
Kategorie: Nádory dětského a adolescentního věku
Téma: 18. Nádory dětí, adolescentů a mladých dospělých
Číslo abstraktu: 167
Autoři: MUDr. Andrea Bobeková; MUDr. Zdeněk Pavelka; doc. MUDr. Leoš Křen, Ph.D.; Mgr. Hana Filková; Mgr. Iva Slámová; Prof.RNDr. Petr Kuglík, CSc.; Doc. MUDr. Jarmila Skotáková, CSc.; MUDr. Jiří Ventruba, CSc.; MUDr. Karel Zitterbart, Ph.D.; prof. MUDr. Jaroslav Štěrba, CSc.
Úvod
Meduloblastomy jsou nejčastější maligní tumory mozku u dětí se závažnou morbiditou a mortalitou [1]. V současnosti se léčebně i prognosticky stratifikují na základě klinických faktorů (věk, přítomnost metastáz, rozsah resekce), histologické varianty (klasická, desmoplastická/nodulární, anaplastická/velkobuněčná) a nověji molekulárních či cytogenetických změn [2]. Prezentujeme kazuistiku pacienta s původně desmoplastickým meduloblastomem, u kterého byla v průběhu onkologické léčby diagnostikována recidiva s konverzí do anaplastické varianty s agresívním biologickým profilem odlišným od původního tumoru.
Kazuistika
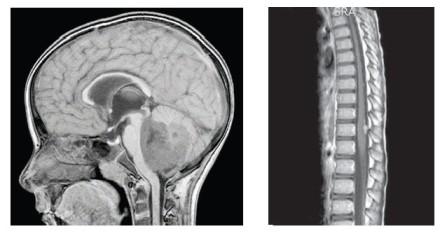
5-letý chlapec s negativní rodinnou anamnézou bez předchozích závažných morbidit se prezentoval příznaky intrakraniální hypertenze, poruchami zrakové ostrosti a nejistou chůzí. MRI mozku prokázala expanzivně se chovající nehomogenní tumor mozečku s nehomogenním sycením po kontrastu a s „polevovými“ míšnímu metastázami, tj. M3 nemoc, vysoké riziko (obr. 1,2). Pacient absolvoval totální exstirpaci primárního tumoru, histologicky byla potvrzena diagnóza meduloblastomu, desmoplastická varianta, bez amplifikace MYCC, MYCN (FISH vyšetření). Morfologický obraz tvořily uzly s buňkami se světlými jádry a s tmavými denzními jádry, polymorfní, mitoticky aktivní. Imunohistochemicky byla prokázána pozitivita synaptophysinu, mitotický index dle Ki67 byl 50 % a více. Zahájena byla léčba pro meduloblastomy vysokého rizika dle modifikovaného protokolu SJMB96 založená na kombinaci konkomitantní chemoradioterapie (temozolomid) s ozářením celé osy, dosycením na metastázy a zadní jámu, následovaly 4 cykly submyeloablativní chemoterapie s podporou autologních periferních kmenových buněk. Vzhledem k vysokému riziku nepříznivého průběhu nemoci jsme léčbu posílili cyklicky intraventrikulárně podávaným etoposidem cestou portu (Ommaya rezervoár). Intenzivní část protokolu pacient ukončil v parciální remisi.
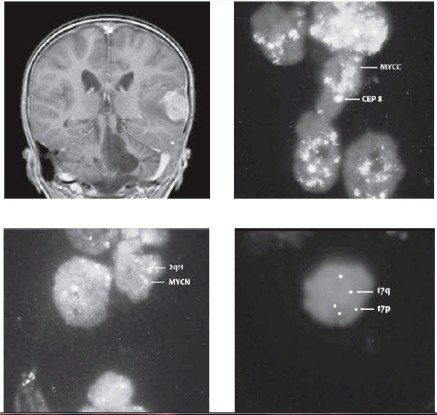
Následovala udržovací léčba kombinací výše uvedeného etoposidu s cykly bioterapie 13-cis retinovou kyselinou. Po 3. cyklu této experimentální léčby MR potvrdilo dosažení 1. kompletní remise onemocnění. Nicméně po krátkém intervalu přichází pacient s příznaky fatické poruchy s váznutím expresivní složky řeči. MRI mozku popsalo recidivu solitární metastázou temporálně vlevo s perifokálním edémem a intenzivním postkontrastním sycením – EFS 13 měsíců (obr. 3). Ložisko bylo radikálně exstirpováno včetně přiléhající tvrdé pleny. Histologicky byla prokázána konverze do anaplastické varianty meduloblastomu s četnými atypickými mitózami, proliferační aktivita dle Ki67 je 70 %. Imunohistochemicky pozitivita synaptophysinu, slabě i chromograninu, fokálně NSE. Tumor změnil i biologické charakteristiky, v metastáze byla potvrzena metodou FISH amplifikace MYCC s více než 50 kopiemi genu, aditivní kopie genu MYCN (3 kopie/2) a izochromosom 17q jako marker delece p53 (obr. 4-6). Operací pacient dosáhl 2. radiologické remise onemocnění. Byla zahájena experimentální paliativní terapie dle protokolu MEMMAT, což je kombinovaná metronomická antiangiogenní léčba (thalidomide, celecoxib, fenofibrate a střídavě nízce dávkovaný perorální etoposid a cyclophosphamide) spolu s intratékalní aplikací etoposidu střídavě s liposomálním cytarabinem.
Diskuze
Meduloblastom je nejčastější maligní tumor CNS u dětí, představuje 15-20 % primárních mozkových tumorů, tvoří 40 % tumorů lokalizovaných u dětí v zadní jámě [1-2]. V době diagnózy se až v třetině případů vyskytují leptomeningeální metastázy. Meduloblastom je biologicky agresivní tumor WHO grade IV. Jedná se o heterogenní jednotku, současná WHO 2007 klasifikace rozlišuje mimo klasický meduloblastom tyto varianty či subvarianty: desmoplastickou/nodulární, s extenzivní nodularitou a anaplastickou/velkobuněčnou (A/LC) [3-5]. Klinické faktory (věk, rozsah resekce, přítomnost či absence metastatické nemoci) jsou klasické hlavní determinanty určující zařazení pacientů do prognosticky rozdílných kategorií standardního a vysokého rizika s potřebou různě intenzivní terapie. Data z klinických studií svědčí pro rozdílné biologické chování jednotlivých patologických variant a také pro vliv genetických či molekulárních abnormit na prognózu [6]. Metastatická nemoc a anaplastická/velkobuněčná varianta jsou asociovány s nízkým PFS (progression-free survival). Nukleární imunoreaktivita pro Beta-katenin, CTNNB1 mutace a monosomie 6 chromozómu byly identifikovány jako prognosticky příznivé faktory [6,7]. MYCC a MYCN amplifikace je asociována s negativní prognózou [7]. Za meduloblastom nízkého rizika lze považovat onemocnění Beta-katenin pozitivní, bez metastáz a bez A/LC fenotypu nebo MYC amplifikace [5-7]. Meduloblastom vysokého rizika je nemoc definovaná jako metastatická, s A/LC genotypem, nebo MYC amplifikací [5-7]. Kategorie nízkého, standardního a vysokého rizika se vzájemně signifikantně liší léčebnými výsledky.
Nejčastější cytogenetickou odchylkou popisovanou u meduloblastomů jsou abnormity 17. chromosomu (7). Nález isochromozomu 17q (i17q) je spojen se ztrátou funkce tumor-supresorového genu p53 lokalizovaného na 17p. Může se vyskytovat u klasické varianty meduloblastomu a dále v asociaci s amplifikací MYCC a/nebo MYCN u anaplastického/velkobuněčného meduloblastomu (2, 7). Průkaz izo17q je spojen s nepříznivým průběhem onemocnění [7].
Zajímavým fenoménem je konverze méně agresivní varianty meduloblastomu do formy agresivnější při progresi onemocnění. V literatuře nejčastěji popisovanou je změna varianty klasické do A/LC varianty. Konverze desmoplastického meduloblastomu v anaplastický/velkobuněčný typ je raritní. Tuto morfologickou proměnu provází současně kumulace nepříznivých biologických a cytogenetických abnormit, nejčastěji izo17q, amplifikace MYCC či MYCN [8].
Závěr
Presentovaná kasuistika dokumentuje nepříznivý průběh meduloblastomu provázený fenoménem konverze původně méně agresivní histologické varianty do varianty agresivnější se současnou kumulací nepříznivých cytogenetických změn. Histologická verifikace radiologické progrese je nutná pro přesnou klasifikaci tumoru stejně jako pro průkaz případných molekulárních terčů pro cílenou terapii. Prognóza části meduloblastomů vysokého rizika v dětském věku zůstává nadále nepříznivá.
Literatura:
- Tonn JCh, Wetphal M, Rutka JT. (eds). Oncology of CNS Tumors: Second edition. Springer. 2010.
- Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011 Apr 10;29(11): 1400-7.
- Louis DN, Ohgaki H, et al. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol (2007) 114:97–109.
- Eberhart ChG, Burger PC. Anaplasia and Grading in Medulloblastomas. Brain Pathol 2003;13:376-385.
- Leonard JR, Cai DX, et al. Large cell/anaplastic medulloblastomas and medullomyoblastomas: clinicopathological and genetic features. J Neurosurg 95:82–88, 2001
- Ellison D. Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathology and Applied Neurobiology (2002), 28, 257–28.
- Taylor MD, Northcott PA, Korshunov A. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2011 Dec 2. [Epub ahead of print]
- Korshunov A, Benner A, Remke M, et al. Accumulation of genomic aberrations during clinical progression of medulloblastoma. Acta Neuropathol. 2008 Oct;116(4):383-90.
Datum přednesení příspěvku: 19. 4. 2012