Konference: 2004 XXVIII. Brněnské onkologické dny a XVIII. Konference pro sestry a laboranty
Kategorie: Nádory dětského a adolescentního věku
Téma: Nádory dětského a adolescentního věku
Číslo abstraktu: 140
Autoři: MUDr. Pavel Mazánek; MUDr. Viera Bajčiová, CSc.; MUDr. Peter Múdry, Ph.D.; prof. MUDr. Jaroslav Štěrba, CSc.
Neuroblastom (NBL) je nejčastějším
extrakraniálním nádorem dětského věku. Jedná se o nádor vycházející
z primitivních buněk sympatického nervového systému a svým chováním
a biologickými vlastnostmi je stále pro mnoho vědců a lékařů
zdrojem úžasu i frustrace.
V České republice onemocní každý rok neuroblastomem přibližně 20 – 30 dětí. Onemocnění je diagnostikováno v 50% u dětí do 2 let věku, v 75% do 4 a v 90% do 10 let věku. Ve věkové skupině do 1 roku je NBL nejčastější malignitou vůbec a incidence neuroblastomu u kojenců je dvojnásobkem incidence akutních leukémií v této věkové skupině. Incidence neuroblastomu v nekroptickém materiálu u dětí do 3 měsíců věku je 1: 259. To je asi 400 krát více, než je klinická incidence. Svědčí to o spontánní involuci či maturaci neuroblastomu u většiny dětí. Je také zajímavé, že u většina dětských pacientů mladších jednoho roku dojde k regresi jejich choroby buď spontánně nebo pouze s minimálními léčebnými zásahy, i když se často může jednat o onemocnění se vzdálenými metastázami. U starších dětí jsou naopak velmi časté případy, kdy i přes použití nejmodernějších a nejintenzivnějších léčebných multimodálních postupů dochází k progresi onemocnění a následně i smrti. Současné poznatky v biologii a genetice neuroblastomu mají velmi silnou prediktivní hodnotu a dovolují nám poměrně přesnou stratifikaci onemocnění neuroblastomem mezi onemocnění nízkého, středního o vysokého rizika (viz. tab.1). To nám dovolí, vzhledem k výrazné genetické i klinické heterogenitě tohoto onemocnění, individuální volbu terapie – od pouhé observace až k velmi agresivní multimodální terapii.
K významným klinickým prognostickým faktorům u neuroblastomu patří především věk dítěte a lokalisace primárního nádoru. Děti mladší mají lepší prognozu (hranicí je 1 rok event. 18 měsíců), adrenální lokalizace je prognosticky nepříznivá, je spojena s častější generalizací do dřeně, naproti tomu mediastinální či pelvická lokalizace je prognosticky příznivější.
Biologickou povahu onemocnění se snaží popsat histopatologická klasifikace nádorové tkáně podle Shimady, ze sérových markerů ji reflektuje hladina ferritinu, neuron-specifické enolázy a LDH, z biochemických markerů dále poměr HVA a VMA (kyseliny homovanilové a vanilmandlové) v moči. V patogenezi neuroblastomu hraje důležitou roli nespočet genetických změn jako aktivace onkogenů, přebytek či ztráta řady alel nebo změny v ploidii nádorových buněk. Biologická povaha onemocnění může být tedy hodnocena také metodikami molekulární genetiky a molekulární cytogenetiky. Jednou z nejdůležitějších genetických změn v genomu neuroblastomových buněk je amplifikace NMYC onkogenu. Amplifikace NMYC onkogenu je spojena především s pokročilými stadii onemocnění, ale i u dětí s nižšími stadii je nález jeho amplifikace spojen s rychlou progresí onemocnění a velmi špatnou prognózou. V současnosti je tento gen prokazován standardně u dě tí s neuroblastomem pomocí metody FISH (fluorescenční in situ hybridizace) (viz obrázek)

Charakteristickou změnou neuroblastomových buněk spojenou s přebytkem genetického materiálu je zmnožení genetického materiálu na dlouhém raménku chromosomu 17 (tzv. 17q gain). Prokázané změny v tomto regionu jsou přítomny přibližně u 50% pacientů a jsou výrazným negativním prognostickým znakem. Další prognosticky významnou změnu v genomu buněk neuroblastomu je delece krátkého raménka chromosomu 1 (del 1p). Tato změna je nalezána asi ve 30% všech onemocnění. Prognostický význam delece 1p byl po dlouhou dobu kontroverzní. Studie z poslední doby však ukazují, že nález del 1p je nezávislý prediktivní faktor pro časnou progresi choroby, ale přitom nemá vliv na délku celkového přežití onemocnění neuroblastomem.
Z řady dalších změn, při kterých dochází ke ztrátě genetického materiálu v buňkách neuroblastomu je i delece 11q. Tato změna je jako nejčastější delece nalézána u téměř 43% pacientů s neuroblastomem. Delece 11q je tak velmi důležitý negativní prediktivní faktor především pro ty pacienty, u kterých v tkáni nádoru nedochází k amplifikaci NMYC onkogen.
Faktory, které jsou odpovědné za regulaci maligní transformaci neuroblastu do buněk vysoce maligního neuroblastomu nejsou doposud zcela objasněny, ale s velkou pravděpodobností jsou tyto děje ovlivňovány neurotrofinovými receptory. Zatímco vysoká exprese TrkA receptoru je charakteristická především pro prognosticky příznivá onemocnění mladších dětí a kojenců a nízká klinická stadia onemocnění při absenci NMYC amplifikace, vysoká exprese receptoru TrkB je prognosticky nepříznivým faktorem. TrkB/BNDF autokrinní stimulace podporuje navíc aktivně angiogenezi a je zodpovědná v určité míře i za lékovou rezistenci neuroblastomu. TrkC na rozdíl od TrkB je podobně jako TrkA specifický pro prognosticky příznivá onemocnění.
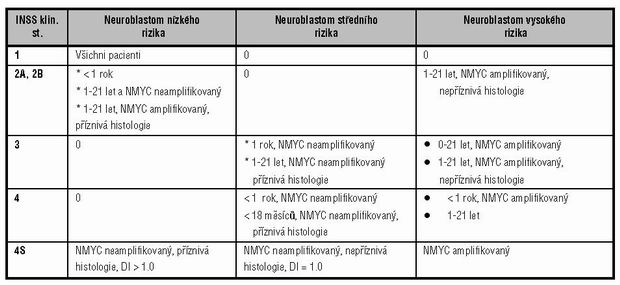
Tabulka:
Terapeutické skupiny onemocnění podle klinických a biologických vlastností neuroblastomu dle INSS
(The International Neuroblastoma Staging System)
Vzhledem ke klinické důležitosti výše uvedených genetických abnormalit u neuroblastomu spektrum genetických vyšetření na KDO FN Brno zahrnuje klasické cytogenetické vyšetření, vyšetření chromozomových abnormalit metodou CGH, průkaz amplifikace NMYC onkogenu metodou FISH a stanovení DNA indexu. V posledním roce bylo spektrum genetických vyšetření u neuroblastomu díky zavedení metody quantitative real-time PCR (qRT-PCR) rozšířeno o kvantitativní vyšetření exprese mRNA neurotrofinových receptorů (TrkA, TrkB a TrkC) a o kvantitativní vyšetření exprese mRNA markerů určených k detekci miniImální reziduální nemoci (kvantifikace exprese mRNA thyrosin hydroxylázy, GD-2 syntházy, PGP 9.5, MAGE 1-6, GAGE a hTERT).
Léčba pacientů s neuroblastomem odráží výraznou klinickou a biologickou heterogenitu tohoto onemocnění. Pro naprostou většinu pacientů s neuroblastomem nízkého rizika je primární terapeutickou modalitou vedoucí k vyléčení chirurgické odstranění tumoru a chemoterapie je u těchto pacientů vyhrazena pro případ rekurence onemocnění, nebo pro případ ohrožení vitálních funkcí nemocného velikostí nebo lokalizací nádoru. U neuroblastomu středního rizika je již k dosažení kontroly nad tímto onemocnění nutné použít mutimodální terapie – konvenční chemoterapii, primární či sekundární resekce nádoru a pooperační radioterapie. V případě neuroblastomu vysokého rizika je často i použití vysocedávkované chemoterapie v kombinaci s ABMT, radioterapií a dlouhodobou udržovací terapií deriváty kyselina retinové nedostatečné. Na základě dlouhodobých zahraničních klinických zkušeností je přežití bez známek choroby 3 roky od stanovení diagnosy u skupiny nízkého rizika 95-100%, středního 85-90% a u pacientů vysokého rizika klesá pod 30%. Ze zkušeností Kliniky dětské onkologie s léčbou neuroblastomů v letech 1998-2003 vyplývá, že z celkového počtu 40 pacientů přežívá nyní 68% pacientů (27/40). Ve skupině neuroblastomu vysokého rizika v současné době přežívá 53% pacientů (10/19), 85% pacientů s neuroblastomem středního rizika (11/13) a 100% pacientů s neuroblastomem nízkého rizika (6/6). Tyto výsledky budou v diskusi srovnány s výše uvedenými molekulárně genetickými vyšetřeními.
Podpořeno VZ FN Brno – MZ 00065269705
V České republice onemocní každý rok neuroblastomem přibližně 20 – 30 dětí. Onemocnění je diagnostikováno v 50% u dětí do 2 let věku, v 75% do 4 a v 90% do 10 let věku. Ve věkové skupině do 1 roku je NBL nejčastější malignitou vůbec a incidence neuroblastomu u kojenců je dvojnásobkem incidence akutních leukémií v této věkové skupině. Incidence neuroblastomu v nekroptickém materiálu u dětí do 3 měsíců věku je 1: 259. To je asi 400 krát více, než je klinická incidence. Svědčí to o spontánní involuci či maturaci neuroblastomu u většiny dětí. Je také zajímavé, že u většina dětských pacientů mladších jednoho roku dojde k regresi jejich choroby buď spontánně nebo pouze s minimálními léčebnými zásahy, i když se často může jednat o onemocnění se vzdálenými metastázami. U starších dětí jsou naopak velmi časté případy, kdy i přes použití nejmodernějších a nejintenzivnějších léčebných multimodálních postupů dochází k progresi onemocnění a následně i smrti. Současné poznatky v biologii a genetice neuroblastomu mají velmi silnou prediktivní hodnotu a dovolují nám poměrně přesnou stratifikaci onemocnění neuroblastomem mezi onemocnění nízkého, středního o vysokého rizika (viz. tab.1). To nám dovolí, vzhledem k výrazné genetické i klinické heterogenitě tohoto onemocnění, individuální volbu terapie – od pouhé observace až k velmi agresivní multimodální terapii.
K významným klinickým prognostickým faktorům u neuroblastomu patří především věk dítěte a lokalisace primárního nádoru. Děti mladší mají lepší prognozu (hranicí je 1 rok event. 18 měsíců), adrenální lokalizace je prognosticky nepříznivá, je spojena s častější generalizací do dřeně, naproti tomu mediastinální či pelvická lokalizace je prognosticky příznivější.
Biologickou povahu onemocnění se snaží popsat histopatologická klasifikace nádorové tkáně podle Shimady, ze sérových markerů ji reflektuje hladina ferritinu, neuron-specifické enolázy a LDH, z biochemických markerů dále poměr HVA a VMA (kyseliny homovanilové a vanilmandlové) v moči. V patogenezi neuroblastomu hraje důležitou roli nespočet genetických změn jako aktivace onkogenů, přebytek či ztráta řady alel nebo změny v ploidii nádorových buněk. Biologická povaha onemocnění může být tedy hodnocena také metodikami molekulární genetiky a molekulární cytogenetiky. Jednou z nejdůležitějších genetických změn v genomu neuroblastomových buněk je amplifikace NMYC onkogenu. Amplifikace NMYC onkogenu je spojena především s pokročilými stadii onemocnění, ale i u dětí s nižšími stadii je nález jeho amplifikace spojen s rychlou progresí onemocnění a velmi špatnou prognózou. V současnosti je tento gen prokazován standardně u dě tí s neuroblastomem pomocí metody FISH (fluorescenční in situ hybridizace) (viz obrázek)
Charakteristickou změnou neuroblastomových buněk spojenou s přebytkem genetického materiálu je zmnožení genetického materiálu na dlouhém raménku chromosomu 17 (tzv. 17q gain). Prokázané změny v tomto regionu jsou přítomny přibližně u 50% pacientů a jsou výrazným negativním prognostickým znakem. Další prognosticky významnou změnu v genomu buněk neuroblastomu je delece krátkého raménka chromosomu 1 (del 1p). Tato změna je nalezána asi ve 30% všech onemocnění. Prognostický význam delece 1p byl po dlouhou dobu kontroverzní. Studie z poslední doby však ukazují, že nález del 1p je nezávislý prediktivní faktor pro časnou progresi choroby, ale přitom nemá vliv na délku celkového přežití onemocnění neuroblastomem.
Z řady dalších změn, při kterých dochází ke ztrátě genetického materiálu v buňkách neuroblastomu je i delece 11q. Tato změna je jako nejčastější delece nalézána u téměř 43% pacientů s neuroblastomem. Delece 11q je tak velmi důležitý negativní prediktivní faktor především pro ty pacienty, u kterých v tkáni nádoru nedochází k amplifikaci NMYC onkogen.
Faktory, které jsou odpovědné za regulaci maligní transformaci neuroblastu do buněk vysoce maligního neuroblastomu nejsou doposud zcela objasněny, ale s velkou pravděpodobností jsou tyto děje ovlivňovány neurotrofinovými receptory. Zatímco vysoká exprese TrkA receptoru je charakteristická především pro prognosticky příznivá onemocnění mladších dětí a kojenců a nízká klinická stadia onemocnění při absenci NMYC amplifikace, vysoká exprese receptoru TrkB je prognosticky nepříznivým faktorem. TrkB/BNDF autokrinní stimulace podporuje navíc aktivně angiogenezi a je zodpovědná v určité míře i za lékovou rezistenci neuroblastomu. TrkC na rozdíl od TrkB je podobně jako TrkA specifický pro prognosticky příznivá onemocnění.
Tabulka:
Terapeutické skupiny onemocnění podle klinických a biologických vlastností neuroblastomu dle INSS
(The International Neuroblastoma Staging System)
Vzhledem ke klinické důležitosti výše uvedených genetických abnormalit u neuroblastomu spektrum genetických vyšetření na KDO FN Brno zahrnuje klasické cytogenetické vyšetření, vyšetření chromozomových abnormalit metodou CGH, průkaz amplifikace NMYC onkogenu metodou FISH a stanovení DNA indexu. V posledním roce bylo spektrum genetických vyšetření u neuroblastomu díky zavedení metody quantitative real-time PCR (qRT-PCR) rozšířeno o kvantitativní vyšetření exprese mRNA neurotrofinových receptorů (TrkA, TrkB a TrkC) a o kvantitativní vyšetření exprese mRNA markerů určených k detekci miniImální reziduální nemoci (kvantifikace exprese mRNA thyrosin hydroxylázy, GD-2 syntházy, PGP 9.5, MAGE 1-6, GAGE a hTERT).
Léčba pacientů s neuroblastomem odráží výraznou klinickou a biologickou heterogenitu tohoto onemocnění. Pro naprostou většinu pacientů s neuroblastomem nízkého rizika je primární terapeutickou modalitou vedoucí k vyléčení chirurgické odstranění tumoru a chemoterapie je u těchto pacientů vyhrazena pro případ rekurence onemocnění, nebo pro případ ohrožení vitálních funkcí nemocného velikostí nebo lokalizací nádoru. U neuroblastomu středního rizika je již k dosažení kontroly nad tímto onemocnění nutné použít mutimodální terapie – konvenční chemoterapii, primární či sekundární resekce nádoru a pooperační radioterapie. V případě neuroblastomu vysokého rizika je často i použití vysocedávkované chemoterapie v kombinaci s ABMT, radioterapií a dlouhodobou udržovací terapií deriváty kyselina retinové nedostatečné. Na základě dlouhodobých zahraničních klinických zkušeností je přežití bez známek choroby 3 roky od stanovení diagnosy u skupiny nízkého rizika 95-100%, středního 85-90% a u pacientů vysokého rizika klesá pod 30%. Ze zkušeností Kliniky dětské onkologie s léčbou neuroblastomů v letech 1998-2003 vyplývá, že z celkového počtu 40 pacientů přežívá nyní 68% pacientů (27/40). Ve skupině neuroblastomu vysokého rizika v současné době přežívá 53% pacientů (10/19), 85% pacientů s neuroblastomem středního rizika (11/13) a 100% pacientů s neuroblastomem nízkého rizika (6/6). Tyto výsledky budou v diskusi srovnány s výše uvedenými molekulárně genetickými vyšetřeními.
Podpořeno VZ FN Brno – MZ 00065269705
Datum přednesení příspěvku: 26. 5. 2004