Konference: 2011 2. pražské mezioborové onkologické kolokvium
Kategorie: Zhoubné nádory plic a průdušek
Téma: Prezentace
Číslo abstraktu: 006
Autoři: MUDr. Filip Janků; Ignacio Garrido-Laguna; prof. MUDr. Luboš Petruželka, CSc.; D.J. Stewart; Dr. Razelle Kurzrock
Abstract
The development of personalized medicine with a focus on novel
targeted therapies has supplanted the one-size-fits-all approach to
the treatment of many cancers, including non-small cell lung cancer
(NSCLC). Targeted therapies, if given to a patient subpopulation
enriched by the presence of relevant molecular targets, can often
abrogate cell signaling that perpetuates cancer progression.
Critical targets activating pro-cancer pathways include,but are not
limited to, epidermal growth factor receptor (EGFR),MET, vascular
endothelial growth factor (VEGF), VEGF receptor (VEGFR), KRAS,
HER2, EML4-ALK, PIK3CA, and insulin-like growth factor 1 receptor
(IGF-1R). Some target-directed therapies, such as EGFR tyrosine
kinase inhibitors and anti-VEGF monoclonal antibody, have already
been approved for clinical use.
Others, such as those targeted to MET, VEGFR, EML4-ALK, and IGF-1R,
are in clinical testing. This review describes molecular targets in
NSCLC that are in development or being clinically applied and their
implications for developing novel anticancer therapies for this
previously refractory malignancy.
Introduction
The treatment outcomes in advanced/metastatic non-small cell lung
cancer (NSCLC) remain unsatisfactory. Standard-of-care palliative
chemotherapeutic regimens only modestly prolong survival. (1,
2) Major discoveries in the molecular biology of human
malignancies has led to successful application of targeted
therapeutic strategies in several cancers. (3-6)
Substantial progress has been made towards understanding the tumor
biology of NSCLC during the last five years.
Epidermal receptor growth factor (EGFR) tyrosine kinase inhibitors
(TKIs) (erlotinib) and antibody targeting vascular endothelial
growth factor (VEGF) (bevacizumab) have been approved for clinical
use. (7, 8) In addition, other therapies targeting EGFR,
MET, VEGF receptor VEGFR), EML4-ALK, HER2, insulin-like growth
factor 1 receptor (IGF-1R), and others are in clinical testing.
(9-11) Furthermore, the first results of large-scale
efforts investigating relevant molecular aberrations have been
recently published(12, 13) This article delineates the
role of molecular targets in NSCLC that are in development or being
clinically applied.
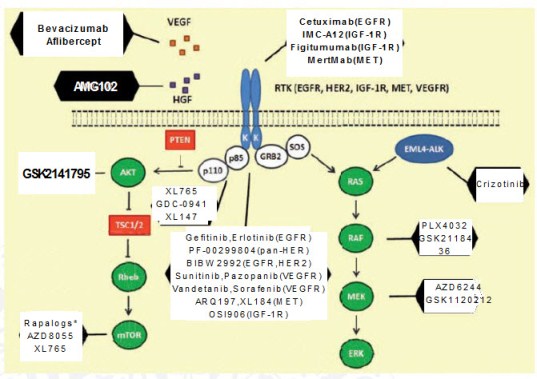
EGFR
EGFR is instrumental in activating the PBK/AKT/mTOR and
RAS/RAF/MEK/MAPK signaling pathways in solid tumors (Figure 1).
(14) These pathways are important in tumor cell growth,
local invasion, angiogenesis, protein translation, autophagy, and
cell metabolism. EGFR gene amplification is present in 30% to
60%(15-17) of NSCLC cases. Furthermore EGFR mutations in
the tyrosine kinase domain are found in approximately 10% of
Caucasian patients and in more than 30% of East Asian patients with
NSCLC (Table 1). (18-21) EGFR has been clinically
investigated for more than a decade as a potential target for
anticancer therapy. Therapeutic strategies for targeting EGFR
include TKIs and monoclonal antibodies.
EGFR tyrosine kinase inhibitors alone
Two principal EGFR TKIs, erlotinib and gefitinib,demonstrated
promising clinical activity. (7, 22) Erlotinib, gained
Food and Drug Administration (FDA) and European Medicines Agency
(EMEA) approval based on the results of a pivotal international
phase III trial, which demonstrated a survival advantage compared
to placebo (6.7 months versus 4.7 months, hazard ratio [HR] 0.70, p
< 0.001) in previously treated patients with advanced NSCLC
(Table 2). (7) In a retrospective subanalysis, erlotinib
yielded better results in women, never-smokers, and in patients
with adenocarcinomas. However, there was a 2.1 month overall
survival (OS) benefit in the same retrospective analysis, even in
the subgroup of male smokers with squamous-cell carcinoma, who
would theoretically be the most disadvantaged
population.(23) Tissue samples were available from 24%
of patients for EGFR mutation analysis. Patients who had tumors
with classic EGFR-activating mutations had a hazard ratio (HR) for
death of 0.65, but this was not statistically significant which may
have been attributed to the small number of patients assessed, as
only 19 patients had EGFR activating mutations.
A trial testing gefitinib versus placebo in previously treated
patients with advanced NSCLC showed no survival advantage despite
the robust sample size (n = 1.692) (Table 2).(24) Median
survival trended toward favoring gefitinib (5.6 months versus 5.1
months, p = 0.087), and time to treatment failure was longer in the
gefitinib group than in the placebo group (3.0 versus 2.6 months, p
= 0.0006) (Table 2). Preplanned subgroup analyses demonstrated a
significantly longer survival in the gefitinib group than in the
placebo group for patients who would be anticipated to have an
increased rate of EGFR-activating mutations, including
never-smokers (median survival 8.9 versus 6.1 months, p = 0.012),
and in patients of East Asian origin (median survival 9.5 versus
5.5 months,p = 0.01). Similar to the erlotinib study, other
retrospective efforts have attempted to identify possible
predictors of better outcomes in NSCLC. (15) However, in
this study, tissue samples were only available in 27 % of cases.
EGFR mutations were detected in 12% of tested samples. In this
study, a striking difference in the response rate (RR) to gefitinib
was observed in patients harboring EGFR mutations compared to
patients with wild-type EGFR (37.5 % versus 2.6 %). Unfortunately,
the responding subgroup was too small to be evaluated for
survival.
Why phase III studies showed improved OS only for erlotinib and not
for gefitinib remains to be fully understood, particularly in view
of various other positive study findings. While erlotinib may, in
fact, be superior to gefitinib, the two agents have not been
compared directly with each other, so that there are no data to
support a true difference between their efficacies. Furthermore,
the RRs (8.9 % for erlotinib and 8 % for gefitinib) were similar
for the two agents in both randomized studies, and the median time
to failure for gefitinib (3 months) was somewhat longer than the
median progression-free survival (PFS) with erlotinib (2.2 months)
(Table 2).(7, 24) A second possibility is that
subsequent therapy had a role in OS. Both agents were associated
with small, but statistically significant prolongations of PFS or
time to treatment failure. A third possibility may reside in the
diverse patient populations being treated. Simulations have
demonstrated that if an agent works only in a small subpopulation
of patients exhibiting a drug target, and not in populations that
do not exhibit the target, then very small differences in the
proportions of patients expressing the target can determine whether
an agent will or will not have a significant impact on
survival.(25) Additionally, patients treated on the
erlotinib study demonstrated a higher median RR to prior therapy
than patients treated on the gefitinib trial (38 % for erlotinib
versus 18 % for gefitinib), which suggests the possibility of
causative differences between the two study populations themselves.
Importantly, erlotinib, unlike gefitinib, was administered at the
maximum tolerated dose and was associated with a higher probability
of patients developing a rash than gefitinib. It is well documented
that the efficacy of EGFR TKIs correlates directly with rash
severity.(26) Despite the fact that higher doses of
gefitinib did not show better efficacy in earlier phase II
trials,(27, 28)the possibility that gefitinib was dosed
suboptimally in the phase III population cannot be completely
disregarded.
The hypothesis that gefitinib is active in patients harboring EGFR
mutations was tested in a small phase II trial.(29)In
total, 30 EGFR mutation-positive patients were enrolled, including
22 with a poor Eastern Cooperative Oncology Group performance
status of 3 or 4. The overall RR was 66% and the total disease
control (complete response, partial response, stable disease) rate
was 90 %. The performance status improvement rate was 79 %, and 68
% of the 22 patients improved from performance status 3 at baseline
to performance status 1. Median PFS reached 6.5 months and median
OS 17.8 months. These observations were confirmed in another phase
II trial, which enrolled 31 patients with advanced NSCLC and
underlying EGFR mutations. (30) Only two patients
progressed on therapy, demonstrating an overall RR of 55 %, median
PFS of 9.2 months, and projected OS of 17.5 months. Of the two
patients who progressed, one had MET amplification, in keeping with
the preclinical observation that MET amplification confers
resistance to EGFR TKIs. (31) The Spanish Lung Cancer
Group screened 2.105 chemonaive or previously treated patients with
NSCLC and found EGFR mutations in 350 patients (16.6 %). Of these
350 patients, 217 were treated with erlotinib, yielding an overall
RR of 70.6 %, median PFS of 14 months and median OS of 27 months.
(19)
In the first-line setting, Mok et al. (32) carried out a
large phase III randomized trial comparing gefitinib with standard
carboplatin/paclitaxel chemotherapy in chemonaive East Asian
patients with advanced lung adenocarcinoma who had a limited
history of smoking (never-smokers and ex-light-smokers). The trial
was designed as a noninferiority study in which the patient
population was enriched by inclusion criteria to increase the
likelihood of response to gefitinib based on retrospective
observations from earlier studies. (7, 24) More than
1.200 patients were randomized (Table 2). The trial exceeded its
primary endpoint (noninferiority), showing in the intent-to-treat
population a 26% risk reduction of disease progression with
gefitinib (HR 0.74, 95 % CI 0.65-0.85,p < 0.001). Gefitinib also
achieved a greater RR compared with carboplatin/paclitaxel (43 %
versus 32.2%, p < 0.001). OS was comparable between the two
arms, but the patients on gefitinib had a better quality of life as
assessed using the FACT-L questionnaire. Tissue samples were
available in 56.1 % of cases. EGFR was mutated in 59.7 % patients
who had available tissue samples and those patients had a 71.2 % RR
with gefitinib compared to 47.3 % for patients treated with
chemotherapy. In contrast, only 1.1 % of patients with no EGFR
mutation responded to gefitinib, whereas 23.5 % responded to
chemotherapy. Similarly, PFS was longer in the gefitinib arm
compared with chemotherapy in patients with EGFR mutations (HR
0.48, 95% CI 0.36-0.64, p < 0.001). The opposite was true for
patients without mutations (HR 2.85, 95% CI 2.05-3.98, p <
0.001). On the basis of these data, gefitinib was approved by the
EMEA as a first-line treatment for patients with advanced or
metastatic NSCLC with underlying EGFR mutations. PFS and RR data
based on specific EGFR mutations were used for post hoc analysis.
(33) The subgroup of 66 patients with an exon 19
deletion had a significantly prolonged PFS following gefitinib
treatment compared with chemotherapy, as reflected by a HR of 0.377
(95% CI 0.255-0.560). RR data showed that gefitinib produced a
response in 84 % of patients and chemotherapy produced responses in
43.2 % of patients. In 64 patients who had an exon 21 L858R
mutation, PFS was increased by gefitinib compared with carboplatin
and paclitaxel, albeit with a less robust HR of 0.553 (95% CI
0.352-0.868) and a smaller difference in RR (gefitinib 60.9 %
versus chemotherapy 53.2 %). FISH subanalysis showed that patients
with a high EGFR copy number and wild-type EGFR did not benefit
from gefitinib, but did benefit from chemotherapy. A limitation of
these results was the small sample size (n = 55).
A smaller phase III trial assessing gefitinib versus carboplatin
and paclitaxel randomly assigned 230 chemonaive patients with EGFR
mutations to receive either gefitinib 250 mg daily or chemotherapy
(Table 2).(34) The median PFS (10.8 months for
gefitinib, 5.4 months for chemotherapy; p < 0.001) and overall
RR results (73.7 % for gefitinib, 30.7 % for chemotherapy; p <
0.001) favored gefitinib. The median OS rates numerically, but not
statistically, also favored gefitinib (30.5 months for gefitinib,
23.6 months for chemotherapy; p = 0.31).
Recently, similar outcomes were replicated with erlotinib, which
was compared to carboplatin and gemcitabine given as first-line
therapy in a randomized phase III trial on 165 patients with
advanced/metastatic NSCLC and activating EGFR mutations (Table 2).
(35) Patients treated with erlotinib demonstrated nearly
tripled PFS of 13.1 months in comparison to median PFS of 4.6
months on chemotherapy (p < 0.0001). OS data are pending.
Prospective and retrospective studies consistently showed overall
better outcomes in patients with EGFR mutations treated both with
EGFR TKIs and chemotherapy. (32, 34, 36, 37) EGFR
mutations are predictive of a high RR and prolonged PFS in patients
treated with anti-EGFR TKIs, which does not, however, translate
into a benefit in OS.
EGFR tyrosine kinase inhibitors as maintenance
therapy
EGFR TKIs are administered orally and have a favorable toxicity
profile, which supports their possible role as NSCLC maintenance
therapy. Erlotinib was tested as a maintenance therapy in
unselected patients with NSCLC without progression after four
cycles of platinum doublets(Table 2)38. Altogether,
1.949 patients were registered at the time of chemotherapy
initiation and 889 were randomly assigned to receive either
erlotinib or placebo. The trial met its primary endpoint,
demonstrating a HR of 0.71 (p < 0.0001) favoring the erlotinib
maintenance arm, however, the difference in median PFS compared to
placebo was only 8.4 days (2.9 months versus 2.6 months). EGFR
mutation analysis was performed in about 40 % of patients. Although
patients with wild-type EGFR had a small PFS benefit from erlotinib
(HR 0.78, p = 0.0185), the most striking difference was observed in
a small patient subpopulation with EGFR mutations (approximately 10
% of the tested samples). Patients with EGFR mutations who were
treated with erlotinib had a HR of 0.10 (p < 0.0001). The
difference in median PFS for the whole study population translated
into a survival advantage (12 months for erlotinib versus 11.1
months for placebo). Interestingly, the survival difference was
maintained even in patients without EGFR mutations (HR 0.77, p =
0.0243). Although this study suggested that patients with EGFR
mutations might derive a meaningful benefit from erlotinib; some
patients with wild-type EGFR derived a small, albeit statistically
significant, PFS and OS benefit from erlotinib maintenance therapy.
Therefore complex genetic or epigenetic changes might be associated
with response to anti-EGFR therapy in the absence of EGFR
mutations. To date, efforts to identify the important changes have
not been conclusive.(39)
EGFR tyrosine kinase inhibitors with
chemotherapy
Erlotinib and gefitinib were each tested in combination with a
standard platinum-containing front-line therapy in four large
randomized trials (Table 2).(40-43) Unfortunately, there
was no improvement in treatment outcomes in any of them. The
disappointing results could be accounted for by various factors.
First, gefitinib and erlotinib were examined in unselected patient
populations. EGFR TKIs are potent drugs in a small proportion of
selected NSCLC patients, but the overall success of these agents in
an unselected patient population is overshadowed by the majority of
patients who are resistant to them. Furthermore, crossover to
second-line EGFR TKIs could have had a role. EGFR TKIs might
antagonize chemotherapy effects by blocking cells in the G1 phase
of the cell cycle, as well as interfere with platinum uptake into
tumor cells, by decreasing membrane uptake transporter
expression(44, 45) Since the effect on membrane
transporters could last for several weeks or longer, EGFR TKIs
could hypothetically impact negatively on the efficacy of
chemotherapy given concurrently with an EGFR TKI, but also
chemotherapy administered two to three months fter discontinuation
of the targeted agent.(46)
Types of EGFR mutations
EGFR activating mutations occur in the region of the ATP-binding
pocket of the tyrosine kinase domain. (47) EGFR
mutations target four exons (18-21) (Table 3). (48, 49)
The most prevalent mutations associated with sensitivity to EGFR
TKIs are in-frame deletions of exon 19 (44 %) and L858R
substitutions in exon 21 (41 %).(50) Nucleotide
substitutions in exon 18 and in-frame deletions of exon 20 are less
frequent (5 % and < 1 %, respectively). Despite dramatic
responses to EGFR TKIs, most patients eventually develop disease
progression in less than a year. (34) A single secondary mutation,
T790M in exon 20, is found in about 50 % of patients relapsing
after initial response to EGFR TKIs. (49, 51) Much less
prevalent is a D761Y secondary mutation of exon 19 associated with
resistance to EGFR TKIs in fewer than 1 % of patients who initially
responded to EGFR TKIs.
EGFR monoclonal antibodies
Cetuximab is a chimeric monoclonal antibody directed against the
extracellular domain of EGFR,
which has activity in a broad spectrum of tumor types, including
lung cancer.(52-54)
Promising results from randomized phase II trials led to initiation
of a study that compared cisplatin and vinorelbine with or without
cetuximab in 1.125 patients with advanced EGFR-overexpressing NSCLC
(Table 2).(55) Although this trial did not show any
difference in PFS, it met its primary endpoint, demonstrating a
modest survival gain of 1.2 months for cetuximab containing arm
(11.3 months versus 10.1 months, HR 0.871, 95% CI 0.762-0.996, p =
0.044). The RR was marginally better in the cetuximab arm (36 %
versus 29 %). Whether this result is clinically relevant is
debatable; however, patients treated with cetuximab who developed a
skin rash within the first 3 weeks of treatment achieved an
impressive 15-month median survival versus 8.8 months in patients
with no rash. (56) Whether this side-effect reflects the
higher efficacy of cetuximab, or whether patients with skin rash
have a better prognosis regardless of a specific treatment regimen,
remains to be seen.
Prespecified subanalysis showed that cetuximab provides bene fit
irrespective of histology. Biomarker analysis reflecting EGFR copy
number on FISH and KRAS mutation showed no significant predictive
value for any of the agents assessed, although the HR favoring
cetuximab was slightly better in patients with EGFR amplification
on FISH.(17)
A similar trial was carried out in the USA in nonselected patients
with stage IIIB/IV NSCLC, which randomized 676 patients (Table 2).
(57) This study did not meet its primary endpoint, as
the difference in PFS of 0.16 months did not reach statistical
significance despite a better RR (25.7 % versus 17.2 %) in the
experimental arm. OS curves were overlapping. Tissue samples for
biomarker subanalysis were available for 225 (33 %) patients. EGFR
expression, EGFR copy number, EGFR mutations, and KRAS mutations
did not influence treatment outcomes. (58)
The only study suggesting a prognostic or predictive value
associated with EGFR FISH status was a phase II trial, SWOG 0342,
which randomized 229 patients to carboplatin and paclitaxel with
concurrent or sequential cetuximab.(16) Biomarker data
were available for 76 patients. Patients with high EGFR copy
numbers had a longer PFS and OS in a retrospective analysis.
Whether this finding reflects a prognostic or predictive value of
EGFR status is difficult to assess, as both treatment arms
contained cetuximab. Biomarker data in studies using cetuximab
consistently showed that, unlike in colorectal
cancer(59) , mutant KRAS does not predict NSCLC
resistance to cetuximab. In addition, neither EGFR mutations nor
EGFR amplification predicted a favorable response to cetuximab,
suggesting that monoclonal antibodies have mechanisms of action
that differ from anti-EGFR TKIs. Cetuximab might interact with
other pathways that have not yet been described and might also
alter non-kinase functions of EGFR, as demonstrated in the
preclinical setting. (60) Additional biomarker studies
are needed to predict the benefit of cetuximab in NSCLC. Another
anti-EGFR monoclonal antibody, fully human panitumumab, is
currently being tested in a randomized phase II clinical
trial.
MET
The MET oncogene encoding a transmembrane receptor with tyrosine
kinase activity was identified nearly three decades ago.
(61) The MET receptor is activated upon binding of the
hepatocyte growth factor (HGF), which is secreted by mesenchymal
cells. In cancer, MET is involved in complex prosurvival
mechanisms, including downstream activation of the RAS/RAF/MEK and
PI3K/AKT pathways (Figure 1).
MET amplification has been reported in 17% of patients with NSCLC
and was associated with a dismal prognosis and resistance to EGFR
TKIs.(30,31,62) Activating mutations in the tyrosine
kinase domain of MET are observed in fewer than 3% of patients
(Table 1).(63-65) Preclinical models have shown that MET
is involved in resistance to angiogenesis inhibitors.
(66) Antiangiogenic drugs induce HIF-1a expression,
which in turn triggers transcription of multiple genes, including
MET. (67) This association provides a rationale for
combining MET inhibitors with EGFR TKIs or antiagiogenic
drugs.
The MET pathway could be inhibited by monoclonal antibodies against
HGF, monoclonal antibodies against a MET receptor, or by MET TKIs.
A recent phase I trial with a fully human anti-HGF monoclonal
antibody AMG 102 in combination with the antiVEGF monoclonal
antibody bevacizumab in patients with advanced solid tumors
demonstrated encouraging activity in NSCLC patients.
(68) An anti-MET receptor monoclonal antibody, PRO143966
(MetMab), is currently being evaluated in a randomized phase II
study in combination with erlotinib. Early results suggest possible
benefit in patients with IHC overexpression of MET. (69)
ARQ 197 is a selective, non-ATP competitive inhibitor of MET
kinase. A phase II randomized trial compared ARQ 197 in combination
with erlotinib to placebo and erlotinib in chemotherapy pretreated,
but EGFR TKI-naive, patients with advanced NSCLC (Table 2). The
trial did not meet its primary endpoint as the increase in median
PFS in the experimental arm was not statistically significant (3.8
months versus 2.3 months; p = 0.23); however, when adjusted for
prespecified factors such as histology, KRAS, and EGFR mutations,
there was a significant 32% risk reduction of disease progression
in the experimental arm (HR 0.68, p < 0.05). (70) Patients with
nonsquamous histology, wild-type EGFR, and mutated KRAS fared best
on the experimental arm. XL184 is an oral inhibitor of MET, VEGFR,
RET, KIT and TIE-2. Preliminary data from an ongoing phase Ib/II
study evaluating the safety and efficacy of XL184 in combination
with erlotinib in patients with advanced NSCLC produced encouraging
results in patients with an EGFR T790M mutation (indicating
resistance to EGFR TKIs) and in patients with MET amplification.
(71)
VEGF AND VEGFR
Bevacizumab
Bevacizumab, a monoclonal antibody against VEGF, increased survival
in NSCLC patients when added to standard carboplatin and paclitaxel
chemotherapy. (8) The pivotal phase III study (ECOG
4599) randomized patients who had advanced NSCLC with a nonsquamous
histology. Adding bevacizumab led to a longer OS of 12.3 versus
10.3 months, a longer PFS of 6.2 versus 4.5 months, and a higher RR
of 35% versus 15% (Table 2). Based on this study, bevacizumab
gained FDA and EMEA approval as first-line therapy for advanced
nonsquamous NSCLC. The AVAiL (Avastin in Lung Cancer) study
evaluated the regimen of cisplatin and gemcitabine with placebo or
in combination with one of two doses of bevacizumab (7.5 mg/kg or
15 mg/kg every 3 weeks). A statistically significant, but
clinically minimal difference of 0.4-0.6 months (p = 0.03, and p =
0.003) in PFS (primary endpoint) was seen when bevacizumab was
added. (72) No dose-effect relationship in the
bevacizumab arms was observed. Nevertheless, the chemotherapy arm
demonstrated a better than expected PFS of 6.2 months. Updated
results revealed no significant difference in OS (13.1, 13.6, and
13.4 months, respectively; (Table 2). (73) More than 60%
of patients treated in this trial received some post-protocol
therapy, which may explain the lack of survival benefit. Data on
post-protocol therapies from the pivotal ECOG 4599 study are not
available, which precludes drawing definitive conclusions. An
additional conundrum is the lack of validated biomarkers that can
identify which patients are likely to benefit from the addition of
bevacizumab. High blood pressure has been associated with prolonged
PFS and OS when bevacizumab was added to chemotherapy in a
retrospective analysis of ECOG 4599. (74) This needs to
be confirmed by other studies with bevacizumab in NSCLC, including
further insight on the downstream effects of VEGF suppression and
hypertension.
VEGFR tyrosine kinase inhibitors
Sunitinib, and sorafenib are TKIs that target multiple kinases,
including the VEGFR, and they have been approved for therapy in
metastatic renal-cell carcinoma, gastrointestinal stromal cancer
(GIST) and hepatocellular carcinoma. (75-77) Both drugs
showed promising results in phase II trials in NSCLC, (78,
79) although a pivotal phase III trial in advanced NSCLC
patients demonstrated no advantage when sorafenib was added to
carboplatin and paclitaxel (Table 2). (80) Currently,
sorafenib is being compared with placebo in the third-line and
fourth-line therapy of NSCLC. The trial should be completed by
2011.
A randomized phase II trial tested a VEGFR/EGFR/RET inhibitor,
vandetanib, as a monotherapy or in combination with paclitaxel and
carboplatin compared with paclitaxel and carboplatin in 181
chemonaive, advanced NSCLC patients (Table 2). (81) The
vandetanib monotherapy arm was stopped early since it was less
effective than chemotherapy. No survival difference was observed
between the two remaining treatment arms; however, vandetanib added
to chemotherapy delayed progression by one week. Vandetanib was
also tested as second-line therapy in combination with docetaxel in
a large phase III trial, which randomized 1.391 patients to either
docetaxel and vandetanib or docetaxel and placebo (Table 2).
(82) Early results from this trial showed a significant
difference in RR favoring vandetanib (17% versus 10%, p <
0.001). The trial met its primary endpoint of PFS, favoring the
experimental arm (4 months versus 3.2 months, HR 0.79, 95% CI
0.70-0.90, p < 0.001). The OS did not significantly differ among
treatment arms (10.6 months for docetaxel and vandetanib versus 10
months with docetaxel and placebo, HR 0.91, 95% CI 0.78-1.07, p =
0.196). Assessment of EGFR mutational analysis suffered from the
relatively small sample size.
Other antiangiogenic treatments, such as VEGFR inhibitors
(cediranib, motesanib, pazopanib) and
vasculature disrupting agents are being tested in numerous phase
III studies.
EML4-ALK
EML4-ALK fusion is a rare abnormality detected in approximately
5-7% of patients with NSCLC( Table 1), (83, 84) a
frequency that doubled (13%) in a population of patients with at
least two of the following characteristics: female gender, Asian
ethnicity, never or light smoking history, and adenocarcinoma
histology. (85) Activated ALK, similar to EGFR/HER2,
might constitutively switch on the RAS/RAF signaling pathway
(Figure 1). Patients with EML4-ALK are more likely to be light or
never-smokers, similar to patients with EGFR mutations, although
there is an association with younger age (median age about 50
years) and, per some authors, with male gender. (85)
EML4-ALK is associated with wild-type EGFR and wild-type KRAS.
(83) Patients with EML4-ALK fusion are resistant to EGFR
TKIs. The response to platinum-based chemotherapy does not,
however, seem to be affected by the presence of the EML4-ALK fusion
gene.
Patients with the EML4-ALK fusion demonstrated an extraordinary
response to the MET and ALK inhibitor crizotinib in a phase I/II
trial. (86)Initially, in the dose-escalation portion of
this trial,a response was seen in a patient with NSCLC and an
underlying ELM4-ALK fusion. In the phase II cohort, 82 NSCLC
patients with ELM4-ALK fusion were assessed. Most of them (59 %)
had been heavily pretreated with at least two prior regimens. Most
patients never smoked (never-smokers 76 %, ex-smokers 23 %, current
smokers 1 %) and all but three had adenocarcinomas. In total, 47
out of 82 patients (57 %) experienced objective responses and PFS
at 6 months was 72%. Only six patients showed disease progression.
This trial reiterated the importance of incorporating prospective
molecular profiling into the selection criteria for early-phase
clinical trials examining targeted therapies. The phase III
clinical trial comparing crizotinib with chemotherapy in a
second-line setting is currently underway.
KRAS
Activating mutations in codons 12 and 13 of the KRAS oncogene occur
in about 10-30 % of lung adenocarcinomas(17, 37, 58, 87,
88) KRAS mutations are usually mutually exclusive to EGFR
mutations and occur early in the development of smoking-related
carcinomas (Table 1).(89) Although it might be assumed
that patients with KRAS-mutated carcinomas (mostly adenocarcinomas)
would be resistant to EGFR TKIs or monoclonal antibodies, similar
to patients with metastatic colorectal cancer and mutated KRAS,
subanalyses of studies contradict this hypothesis in NSCLC and
showed similar survival profiles after gefitinib or cetuximab
treatment, irrespective of KRAS status.(17, 37,
58)Generally, in NSCLC, mutant KRAS seems to be associated
with lower response rates to anti-EGFR therapies than wild-type
EGFR.(15, 31,90-93) Because no available drug blocks
KRAS directly, studies are evaluating other potential targets in
the RAS/RAF/ MEK pathway that function downstream of RAS (Figure
1). Apart from sorafenib, which is a relatively weak RAF inhibitor,
encouraging preclinical data suggest a potential benefit from MEK
inhibitors. (94-96) These compounds are currently in the
early stages of clinical research.
HER2
The HER2 oncogene is a member of the EGFR family, which in addition
includes EGFR, HER3 and HER4. (97) These members encode
transmembrane receptors that drive and regulate cellular functions
of cell proliferation. (98) The HER2 receptor does not
have a known ligand and is putatively activated by homodimerization
with other HER2 receptors or by heterodimerization, preferentially
with either EGFR or HER3.(99) The role of the HER4
receptor in cancer is less defined.(100)HER2 and HER3
heterodimers seem to be more potent than HER2
homodimers.(101) HER2 putatively activates the
PI3K/AKT/mTOR and RAS/RAF/MEK pathways. (98)
HER2 overexpression and amplification have been found in diverse
tumors, including breast, gastric and salivary gland
cancers.(102-104)In breast and gastric cancer, HER2
overexpression or HER2 amplification indicate sensitivity to the
anti-HER2 humanized monoclonal antibody trastuzumab.(105,
106)In NSCLC, HER2 amplification is found in 2-23 % of
patients, however, these patients do not seem to benefit from
anti-HER2 monoclonal antibodies (trastuzumab) or HER2 TKIs
(lapatinib).(107-110) Interestingly, HER2 mutations in
exon 20 of the tyrosine kinase domain were found in 3-10 % of lung
adenocarcinomas (Table 1).(112) The most frequent
mutation is YVMA 776-779 ins, which was associated with
constitutive activation of HER2 kinase, leading to phosphorylation
of downstream effectors such as AKT and MEK.)(113) In
the same preclinical experiments, cell lines with mutated HER2 were
also sensitive to the HER2 tyrosine kinase inhibitor lapatinib. In
NSCLC HER2, EGFR, and KRAS mutations seem to be mutually exclusive.
However, a small proportion of patients has coexisting HER2 and
EGFR mutations. (112, 114) Patients with HER2 mutations
are resistant to anti-EGFR TKIs irrespective of their EGFR mutation
status as pro-growth signals are executed through HER2 kinase.
(113) HER2 receptor provides an attractive target for
anticancer therapy in patients with activating mutations. Several
clinical trials investigating the efficacy of anti-HER2 antibodies
(trastuzumab, MGAH22) and HER2 inhibitors (PF-00299804, BIIW 2992)
are currently underway.
PIK3CA
The PI3K/AKT/mTOR pathway is activated in many cancers,including
NSCLC. This signaling pathway plays an important role in the early
stages of lung cancer development.(115)
Molecular aberrations such as amplification or mutations of the
p110a subunit,PIK3CA, can lead to the activation of the pathway.
PIK3CA amplification has been reported in about 12-17% of patients
with NSCLC. (116, 117) Mutations in the helical or
kinase domain of PIK3CA were reported in 2% to 13% of NSCLC
patients (Table 1). (117-119) Numerous drugs interfere
with this pathway, including PI3K, AKT and mTOR inhibitors. Some of
them,such as rapalogs inhibiting the mTOR Complex 1 (mTORC1),
temsirolimus and everolimus, are already approved by the FDA and
EMEA for other indications. (120, 121) Preclinical data
and early clinical experiments suggest that coexisting KRAS and
PIK3CA mutations may be associated with resistance to PI3tyAKT/mTOR
axis inhibitors. (119, 122) Importantly, these agents
might be active even in the absence of a PIK3CA mutation owing to
frequent alterations at other levels of the PI3K/AKT/mTOR pathway,
such as PTEN loss, AKT mutations, and other alterations. In
addition, activation of PI3K/AKT/mTOR pathway may be involved in
resistance to EGFR TKIs. (123)
BRAF
BRAF mutations in the G-loop (exon 11) or activation segment (exon
15) of its kinase domain have been reported in 3% of patients with
NSCLC.(124) BRAF mutations in lung cancer are early
events in lung cancer tumorigenesis and they are qualitatively
different from V600E BRAF mutations in malignant melanoma, which
may have implications for BRAF inhibitor efficacy. (125)
Preclinical data suggested that BRAF mutations might predict
sensitivity of NSCLC cells to MEK inhibitors (Figure 1).
(126) This hypothesis is now being tested in early
clinical trials, with several MEK inhibitors currently in clinical
development.
IGF-1R
The insulin-like growth factor 1 receptor (IGF-1R) is a promising
novel therapeutic target in NSCLC.(127) In operable NSCLC, an
increased copy number of the IGF-1R gene was associated with a
better prognosis(128) At the cellular level, IGF-1R is
activated upon binding of insulin-like growth factor (IGF).
(129) IGF-1R signaling involves the activation of
various intracellular signaling pathways, including the RAS/RAF/MAP
kinase and PI3K/AKT pathways (Figure 1).
A randomized phase II trial demonstrated a better RR when
figitumumab, a monoclonal antibody against IGF-1R, was added to
standard carboplatin and paclitaxel chemotherapy (54 % versus 42 %,
p < 0.001). (130) The RRs were particularly
encouraging in tumors with a squamous histology, supporting the
hypothesis that deregulation of the IGF-1R pathway may be common in
squamous histology NSCLC. (131) Unfortunately, a
subsequent phase III trial comparing figitumumab plus chemotherapy
versus chemotherapy alone was terminated early for futility.
(132) Another, fully human anti-IGF-1R monoclonal
antibody, cixutumumab, is currently being evaluated in phase II
randomized trials.
Conclusion
The discovery of EGFR mutations revolutionized treatment of NSCLC
and triggered the paradigm shift from a one-size-fits-all to more
personalized approaches. Currently, there are myriad novel
therapeutic targets currently being investigated in laboratory and
clinical experiments. An analysis of the major therapeutic advances
in cancer treatment demonstrated that most breakthroughs, whereby
drugs produce very high response rates in a tumor type, have been
observed in uncommon tumors. (3) Classic examples, among others,
are the use of KIT kinase inhibitors in KIT mutation-positive GIST
and the use of BCR-ABL inhibitors in chronic myelogenous leukemia.
(4,5) These tumor types develop predominantly as a result of a
single molecular aberration; this small repertoire of underlying
single abnormalities may be responsible both for the rare
occurrence of associated tumors and being amenable to treatment
once an appropriate targeted agent is identified. By contrast, it
seems plausible that common tumors, such as NSCLC, are composed of
multiple subsets of disease, each with its own molecular
abnormalities (examples include EGFR-mutation-positivity,
EML4-ALK-positivity, HER2 mutations, and others). Identifying the
relevant molecular subtypes of this heterogeneous disease, and
matching patients with appropriate targeted agents rather than
performing large trials in unselected patients, is crucial if we
are to make headway.(133)
According to this paradigm, recognizing even small subsets of
disease may be critical. For example, HER2 mutations are identified
in 3-10 % of NSCLC patients, yet deregulation of this signal might
underlie resistance to EGFR-directed therapy. 111-113)
It is highly likely that many patients have several abnormalities
driving tumorigenesis and that single targeted agents will be
insufficient for providing meaningful therapeutic results. Indeed,
MET amplification or mutation is detected in 3-17 % of NSCLC
patients, and may augment resistance to anti-EGFR
therapy.(30,62)
To address this issue, clinical trials that combine EGFR and MET
kinase inhibitor therapy have been developed.
In conclusion, chemotherapy improves survival in patients with
advanced NSCLC,although its ultimate success has been limited. New
therapeutic combinations involving targeted therapies and
strategies hold promise for improved treatment outcomes, even in
resistant patients. To achieve this benefit current classification
schemes must be revised to incorporate molecular features in order
to better address the requirements of a targeted therapy approach
within the context of personalized medicine, and enable researchers
to add promising new drugs to their therapeutic
armamentarium while sparing patients without relevant molecular
aberrations from unnecessary treatments.
ACKNOWLEDGEMENTS
We thank to Ms. Joann Aaron for scientific review and editing of
this article.
Disclosure of Conflicts of Interest
None of the authors have any conflict of interest relevant to the
subject of this manuscript.
Table 1: Molecular abnormalities in NSCLC
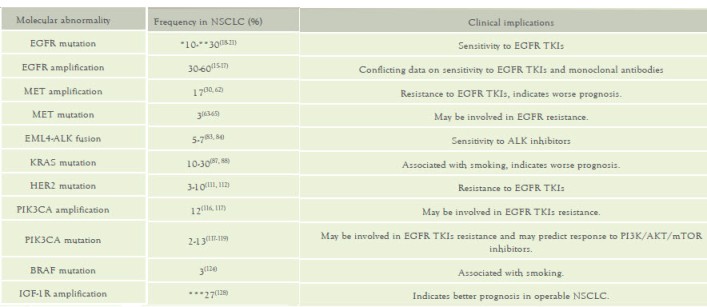
Abbreviations: NSCLC, non-small cell lung cancer; TKIs, tyrosine
kinase inhibitors
* Frequency in Caucasian population
** Frequency in East Asian population
*** Combined high copy number and polysomy
Table 2: Major randomized trials with targeted therapies for
advanced NSCLC
Zvětšení tabulky
Table 3: EGFR mutations
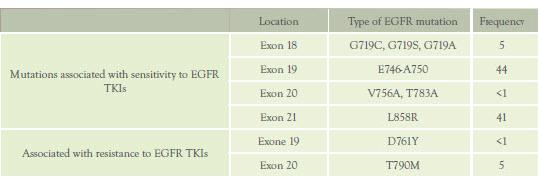
Abbreviations: TKIs; tyrosine kinase inhibitors
Figure 1: Therapeutic targets and novel agents in NSCLC
Janku F.(1), Garrido-Laguna I.(1), Petruzelka L.(2), Stewart DJ(3),
Kurzrock R.(1)
(1)Department of Investigational Cancer Therapeutics (Phase I
Clinical Trials Program), The University of Texas MD Anderson
Cancer Center, Houston, Texas, 1515 Holcombe Blvd., Box 455,
Houston, TX 77030, USA, (2)Department of Oncology, First Faculty of
Medicine, The Charles University Prague, U Nemonice 2, 128 08 Praha
2, Czech Republic, (3)Department of Thoracic/Head & Neck
Medical Oncology, The University of Texas MD Anderson Cancer
Center, 1515 Holcombe Boulevard, Houston, TX 77030, USA.
References
-
Chemotherapy in addition to supportive care improves survival in advanced non-small-cell lung cancer: a systematic review and meta-analysis of individual patient data from 16 randomized controlled trials. J Clin Oncol 2008;26:4617-25.
-
Ellis PA, Smith IE, Hardy JR, et al. Symptom relief with MVP (mitomycin C, vinblastine and cisplatin) chemotherapy in advanced non-small-cell lung cancer. Br J Cancer 1995;71:366-70.
-
Braiteh F, Kurzrock R. Uncommon tumors and exceptional therapies: paradox or paradigm? Mol Cancer Ther 2007;6:1175-9.
-
Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031-7.
-
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472-80.
-
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N Engl J Med 2010;363:809-19.
-
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353:123-32.
-
Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006;355:2542-50.
-
Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359:1367-80.
-
Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer-is it becoming a reality? Nat Rev Clin Oncol 2010;7:401-14.
-
Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol 2010.
-
Weir BA, Woo MS, Getz G, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007;450:893-8.
-
Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008;455:1069-75.
-
Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006;441:424-30.
-
Hirsch FR, Varella-Garcia M, Bunn PA, Jr., et al. Molecular predictors of outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-cell lung cancer. J Clin Oncol 2006;24:5034-42.
-
Hirsch FR, Herbst RS, Olsen C, et al. Increased EGFR gene copy number detected by fluorescent in situ hybridization predicts outcome in non-small-cell lung cancer patients treated with cetuximab and chemotherapy. J Clin Oncol 2008;26:3351-7.
-
O'Byrne KB, Barrios I, Eschbach C, Martens C, Hotko U, Kortsik Y, Celik C, IStroh I, Pirker C. Molecular and clinical predictors of outcome for cetuximab in non-small cell lung cancer (NSCLC): Data from the FLEX study. J Clin Oncol 2009;27:abstr 8007.
-
Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339-46.
-
Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958-67.
-
Mitsudomi T, Kosaka T, Endoh H, et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol 2005;23:2513-20.
-
Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer - molecular and clinical predictors of outcome. N Engl J Med 2005;353:133-44.
-
Kim ES, Hirsh V, Mok T, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet 2008;372:1809-18.
-
Clark GM, Zborowski DM, Santabarbara P, et al. Smoking history and epidermal growth factor receptor expression as predictors of survival benefit from erlotinib for patients with non-small-cell lung cancer in the National Cancer Institute of Canada Clinical Trials Group study BR.21. Clin Lung Cancer 2006;7:389-94.
-
Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005;366:1527-37.
-
Stewart DJ, Whitney SN, Kurzrock R. Equipoise lost: ethics, costs, and the regulation of cancer clinical research. J Clin Oncol 2010;28:2925-35.
-
Wacker B, Nagrani T, Weinberg J, Witt K, Clark G, Cagnoni PJ. Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studies. Clin Cancer Res 2007;13:3913-21.
-
Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol 2003;21:2237-46.
-
Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003;290:2149-58.
-
Inoue A, Kobayashi K, Usui K, et al. First-line gefitinib for patients with advanced non-small-cell lung cancer harboring epidermal growth factor receptor mutations without indication for chemotherapy. J Clin Oncol 2009;27:1394-400.
-
Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 2008;26:2442-9.
-
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43.
-
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947-57.
-
Mok TT, KF. Wu, YL. et al. Clinical outcome of patients with different types of epidermal growth factor receptor mutations in IPASS. J Thorac Oncol 2009;4.
-
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380-8.
-
Zhou C, Wu Y, Chen G, et al. Efficacy results from the randomised phase III Optimal (CTONG 0802) study comparing first-line erlotinib versus carboplatin (CBDCA) plus gemcitabine (GEM), in Chinese advanced non-small-cell lung cancer (NSCLC) patients (PTS) with EGFR activating mutations. Ann Oncol 2010;21:viii6, LBA13.
-
Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005;23:5900-9.
-
Douillard JY, Shepherd FA, Hirsh V, et al. Molecular Predictors of Outcome With Gefitinib and Docetaxel in Previously Treated Non-Small-Cell Lung Cancer: Data From the Randomized Phase III INTEREST Trial. J Clin Oncol 2010;28:744-52.
-
Cappuzzo F, Ciuleanu T, Stelmakh L, et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol 2010;11:521-9.
-
Tan EH, Ramlau R, Pluzanska A, et al. A multicentre phase II gene expression profiling study of putative relationships between tumour biomarkers and clinical response with erlotinib in non-small-cell lung cancer. Ann Oncol;21:217-22.
-
Giaccone G, Herbst RS, Manegold C, et al. Gefitinib in 54. combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial-INTACT 1. J Clin Oncol 2004;22:777-84.
-
Herbst RS, Giaccone G, Schiller JH, et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial-INTACT 2. J Clin Oncol 2004;22:785-94. 56.
-
Gatzemeier U, Pluzanska A, Szczesna A, et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol 2007;25:1545-52.
-
Herbst RS, Prager D, Hermann R, et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol 2005;23:5892-9.
-
Mahaffey CM, Davies AM, Lara PN, Jr., et al. Schedule-dependent apoptosis in K-ras mutant non-small-cell lung cancer cell lines treated with docetaxel and erlotinib: rationale for pharmacodynamic separation. Clin Lung Cancer 2007;8:548-53.
-
Tsai C, Chen T, Chang K, Hsiao S. Combination effects of gefitinib plus cisplatin in non-small cell lung cancer (NSCLC): Why have phase III trials failed? J Clin Oncol 2009;27:abstract 11022.
-
Stewart DJ, Issa JP, Kurzrock R, et al. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res 2009;15:3881-8.
-
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39.
-
Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004;64:8919-23.
-
Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005;352:786-92.
-
Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer 2006;118:257-62.
-
Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res 2006;12:5764-9.
-
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004;351:337-45.
-
Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006;354:567-78.
-
Rosell R, Robinet G, Szczesna A, et al. Randomized phase II study of cetuximab plus cisplatin/vinorelbine compared with cisplatin/vinorelbine alone as first-line therapy in EGFR-expressing advanced non-small-cell lung cancer. Ann Oncol 2008;19:362-9.
-
Pirker R, Pereira JR, Szczesna A, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet 2009;373:1525-31.
-
Gatzemeier U, von Pawel J, Vynnchenko I, et al. FLEX: Cetuximab in combination with platinum-based chemotherapy (CT) improves survival versus CT alone in the 1st-line treatment of patients (pts) with advanced non-small cell lung cancer (NSCLC). J Thorac Oncol 2008;3:abstract 8.
-
Lynch TJ, Patel T, Dreisbach L, et al. Cetuximab and First-Line Taxane/Carboplatin Chemotherapy in Advanced Non-Small-Cell Lung Cancer: Results of the Randomized Multicenter Phase III Trial BMS099. J Clin Oncol 2010;28:911-7.
-
Khambata-Ford S, Harbison CT, Hart LL, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol;28:918-27.
-
Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009;360:1408-17.
-
Weihua Z, Tsan R, Huang WC, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008;13:385-93.
-
Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984;311:29-33.
-
Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 2009;27:1667-74.
-
Forbes SA, Tang G, Bindal N, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res 2010;38:D652-7.
-
Tengs T, Lee JC, Paez JG, et al. A transforming MET mutation discovered in non-small cell lung cancer using microarray-based resequencing. Cancer Lett 2006;239:227-33.
-
Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol 2009;4:5-11.
-
Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009;15:220-31.
-
Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003;3:347-61.
-
Rosen PJ, Sweeney CJ, Park DJ, et al. A phase Ib study of AMG 102 in combination with bevacizumab or motesanib in patients with advanced solid tumors. Clin Cancer Res 2010;16:2677-87.
-
Spigel D, Ervin T, Ramlau R, et al. Randomized multicenter double-blind placebo-controlled phase II study evaluating Metmab, an antibody to MET receptor, in combination with erlotinib, in patients with advanced non-small-cell lung cancer. Ann Oncol 2010;21:viii7, LBA 15.
-
J. H. Schiller, W. L. Akerley, Brugger W, al e. Results from ARQ 197-209: A global randomized placebo-controlled phase II clinical trial of erlotinib plus ARQ 197 versus erlotinib plus placebo in previously treated EGFR inhibitor-naive patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) J Clin Oncol 28:18s, 2010.
-
Wakelee HA, Gettinger SN, Engelman JA, et al. A phase Ib/II study of XL184 (BMS 907351) with and without erlotinib (E) in patients (pts) with non-small cell lung cancer (NSCLC). J Clin Oncol 2010;28:abstr 3017.
-
Reck M, von Pawel J, Zatloukal P, et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAil. J Clin Oncol 2009;27:1227-34.
-
Reck M, von Pawel J, Zatloukal P, et al. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL). Ann Oncol 2010;21:1804-9.
-
Dahlberg SE, Sandler AB, Brahmer JR Schiller JH, Johnson DH. Clinical Course of Advanced Non-Small-Cell Lung Cancer Patients Experiencing Hypertension During Treatment With Bevacizumab in Combination With Carboplatin and Paclitaxel on ECOG 4599. J Clin Oncol.
-
Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125-34.
-
Motzer RJ, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol 2006;24:5601-8.
-
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
-
Socinski MN, S. Sanchez, JM. et al. Efficacy and safety of sunitinib in previously treated, advanced non-small cell lung cancer (NSCLC): Preliminary results of a multicenter phase II trial. J Clin Oncol 2006;24:7001.
-
Gatzemeier UB, G. Fosella, F. Phase II trial of single-agent sorafenib in patients with advanced non-small cell lung carcinoma. J Clin Oncol 2006;24:abstr 7002.
-
Scagliotti G, Novello S, von Pawel J, et al. Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol;28:1835-42.
-
Heymach JV, Paz-Ares L, De Braud F, et al. Randomized phase II study of vandetanib alone or with paclitaxel and carboplatin as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol 2008;26:5407-15.
-
Herbst RS, Y. Korfee, S. Germonpre, P. Saijo, N. Zhou, C. Wang, J. Langmuir, P. Kennedy, SJ. Johnson, BE. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small cell lung cancer (NSCLC): A randomized, double-blind phase III trial (ZODIAC). J Clin Oncol 2009;27:CRA8003.
-
Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009;115:1723-33.
-
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-6.
-
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247-53.
-
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-703.
-
Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med 1987;317:929-35.
-
Mitsudomi T, Steinberg SM, Oie HK, et al. ras gene mutations in non-small cell lung cancers are associated with shortened survival irrespective of treatment intent. Cancer Res 1991;51:4999-5002.
-
Ahrendt SA, Decker PA, Alawi EA, et al. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer 2001;92:1525-30.
-
Douillard JH, V. Mok, TS. Socinski, MA. Watkins, C. Lowe, E. Armour, A. Kim, ES. Molecular and clinical subgroup analyses from a phase III trial comparing gefitinib with docetaxel in previously treated non-small cell lung cancer (INTEREST). J Clin Oncol 2008;26:abstr 8001.
-
Jackman DS, LV. Cioffredi, L. Gallegos Ruiz, M. Janne, 104. PA. Giaccone, G. Miller, VA. Johnson, BE. Impact of EGFR and KRAS genotype on outcomes in a clinical trial registry of NSCLC patients initially treated with erlotinib or gefitinib. J Clin Oncol 2008;26:abstr 8035.
-
Massarelli E, Varella-Garcia M, Tang X, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res 2007;13:2890-6.
-
Zhu CQ, da Cunha Santos G, Ding K, et al. Role of KRAS 107. and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol 2008;26:4268-75.
-
Balko JM, Jones BR, Coakley VL, Black EP. Combined 108. MEK and EGFR inhibition demonstrates synergistic activity in EGFR-dependent NSCLC. Cancer Biol Ther 2009;8.
-
Mahoney CL, Choudhury B, Davies H, et al. LKB1/KRAS mutant lung cancers constitute a genetic subset of NSCLC with increased sensitivity to MAPK and mTOR signalling inhibition. Br J Cancer 2009;100:370-5.
-
Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14:1351-6.
-
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005;5:341-54.
-
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127-37.
-
Garrett TP, McKern NM, Lou M, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell 2003;11:495-505.
-
Feng SM, Sartor CI, Hunter D, et al. The HER4 cytoplasmic domain, but not its C terminus, inhibits mammary cell proliferation. Mol Endocrinol 2007;21:1861-76.
-
Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 2009;9:463-75.
-
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987;235:177-82.
-
Jaehne J, Urmacher C, Thaler HT, Friedlander-Klar H, Cordon-Cardo C, Meyer HJ. Expression of Her2/neu oncogene product p185 in correlation to clinicopathological and prognostic factors of gastric carcinoma. J Cancer Res Clin Oncol 1992;118:474-9.
-
Cornolti G, Ungari M, Morassi ML, et al. Amplification and overexpression of HER2/neu gene and HER2/neu protein in salivary duct carcinoma of the parotid gland. Arch Otolaryngol Head Neck Surg 2007;133:1031-6.
-
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783-92.
-
Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687-97.
-
Cappuzzo F, Varella-Garcia M, Shigematsu H, et al. Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. J Clin Oncol 2005;23:5007-18.
-
Tan D, Deeb G, Wang J, et al. HER-2/neu protein expression and gene alteration in stage I-IIIA non-small-cell lung cancer: a study of 140 cases using a combination of high throughput tissue microarray, immunohistochemistry, and fluorescent in situ hybridization. Diagn Mol Pathol 2003;12:201-11.
-
Pellegrini C, Falleni M, Marchetti A, et al. HER-2/Neu alterations in non-smal cell lung cancer: a comprehensive evaluation by real time reverse transcription-PCR, fluorescence in situ hybridization, and immunohistochemistry. Clin Cancer Res 2003;9:3645-52.
-
Ross HJ, Blumenschein GR, Jr., Aisner J, et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin Cancer Res;16:1938-49.
-
Stephens P, Hunter C, Bignell G, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 2004;431:525-6.
-
Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 2005;65:1642-6.
-
Wang SE, Narasanna A, Perez-Torres M, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006;10:25-38.
-
Cappuzzo F, Ligorio C, Janne PA, et al. Prospective study of gefitinib in epidermal growth factor receptor fluorescence in situ hybridization-positive/phospho-Akt-positive or never smoker patients ith advanced non-small-cell lung cancer: the ONCOBELL trial. J Clin Oncol 2007;25:2248-55.
-
West KA, Linnoila IR, Belinsky SA, Harris CC, Dennis PA. Tobacco carcinogen-induced cellular transformation increases activation of the phosphatidylinositol 3'-kinase/Akt pathway in vitro and in vivo. Cancer Res 2004;64:446-51.
-
Kawano O, Sasaki H, Okuda K, et al. PIK3CA gene amplification in Japanese non-small cell lung cancer. Lung Cancer 2007;58:159-60.
-
Yamamoto H, Shigematsu H, Nomura M, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 2008;68:6913-21.
-
Kawano O, Sasaki H, Endo K, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer 2006;54:209-15.
-
Janku F, Tsimberidou A, Garrido-Laguna I, et al. PIK3CA, KRAS, and BRAF mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. J Clin Oncol 2010;28:abstr 2583.
-
Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356:2271-81.
-
Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449-56.
-
Ihle NT, Lemos R, Jr., Wipf P, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res 2009;69:143-50.
-
Ihle NT, Paine-Murrieta G, Berggren MI, et al. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther 2005;4:1349-57.
-
Brose MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 2002;62:6997-7000.
-
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809-19.
-
Pratilas CA, Hanrahan AJ, Halilovic E, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res 2008;68:9375-83.
-
Ouban A, Muraca P, Yeatman T, Coppola D. Expression and distribution of insulin-like growth factor-1 receptor in human carcinomas. Hum Pathol 2003;34:803-8.
-
Dziadziuszko R, Merrick DT, Witta SE, et al. Insulin-like growth factor receptor 1 (IGF1R) gene copy number is associated with survival in operable non-small-cell lung cancer: a comparison between IGF1R fluorescent in situ hybridization, protein expression, and mRNA expression. J Clin Oncol;28:2174-80.
-
Werner H, Le Roith D. New concepts in regulation and function of the insulin-like growth factors: implications for understanding normal growth and neoplasia. Cell Mol Life Sci 2000;57:932-42.
-
Karp DD, Paz-Ares LG, Novello S, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol 2009;27:2516-22.
-
Han JY, Choi BG, Choi JY, Lee SY, Ju SY. The prognostic significance of pretreatment plasma levels of insulin-like growth factor (IGF)-1, IGF-2, and IGF binding protein-3 in patients with advanced non-small cell lung cancer. Lung Cancer 2006;54:227-34.
-
Jassem J, Langer CJ, Karp DD, et al. Randomized, open label, phase III trial of figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin in patients with non-small cell lung cancer (NSCLC). J Clin Oncol 2010;28:abstr 7500.
-
Stewart DJ, Kurzrock R. Cancer: the road to Amiens. J Clin Oncol 2009;27:328-33.
Datum přednesení příspěvku: 28. 1. 2011