Konference: 2014 19th Congress of the European Hematology Association - účast ČR
Kategorie: Maligní lymfomy a leukémie
Téma: Chronic lymphocytic leukemia and related disorders - Biology (Poster)
Číslo abstraktu: P852
Autoři: Lesley-Ann Sutton; Viktor Ljungström; Larry Mansouri; Emma Young; Diego Cortese; Mgr. Veronika Navrkalová, Ph.D.; RNDr. Jitka Malčíková, Ph.D.; Alice F. Muggen, PhD; Doc. MUDr. Martin Trbušek, PhD; MD Frederic Davi, PhD; MD Chrysoula Belessi; Dr. Anton W. (Ton) Langerak ; M.D. Paolo Ghia, Ph.D.; prof. RNDr. Šárka Pospíšilová, Ph.D.; MD Kostas Stamatopoulos; Prof. MD Richard Rosenquist (Brandell), PhD
ABSSUB-5489
Background: Recently, next generation sequencing (NGS) studies have revealed a number of novel recurrent mutations in CLL, the prime examples being mutations within NOTCH1, SF3B1 and BIRC3, with higher frequencies in patients with a more aggressive disease and a poor clinical outcome. Considering the increasing number of prognostically relevant genes and the relatively high cost and laborious nature of Sanger sequencing, we aimed at exploring targeted NGS as a novel strategy to assess the mutation status of several genes with prognostic potential. Major advantages with this approach revolve around its capacity to screen a large number of genes and patient samples simultaneously (currently up to 96 samples). Aside from the ability to multiplex both genes and patient samples, the high sequence depth achievable also enables tracking minor subclonal populations over time. Since subclonal dynamics are partially shaped by the treatment regimen received, this will become important as new therapies enter the clinic. An additional favorable attribute of targeted NGS is the ability to analyze all coding exons within a gene regardless of length. This is noteworthy since the size of several prognostic genes, such as ATM, would hinder comprehensive gene analysis within a clinical setting.
Aims: To this end, we designed a HaloPlex gene panel (Agilent Technologies) focusing on 10 genes (mean coverage 99%); TP53, ATM, SF3B1, NOTCH1, BIRC3, MYD88 and POT1 have been linked to CLL prognosis, while XPO1, KLHL6, LRP1B are less characterized but were observed in various NGS studies.
Methods: A total of 168 high risk CLL patients were investigated (unmutated IGHV, n=119; IGHV3-21 subset #2, n=49). Sequencing libraries were run on the Illumina 2000 HiSeq instrument and a mean read depth of ~1500 reads/base within the regions of interest was obtained. Data were analyzed using our in-house bioinformatics pipeline that required the following conditions to be met for a variant to pass all filtering steps: (i) exonic or in a splicing region; (ii) non-synonymous or resulting in a frameshift; (iii) not listed in dbSNP137; (iv) variant allele frequency > 0.1; and, (v) variant allele read depth > 10.
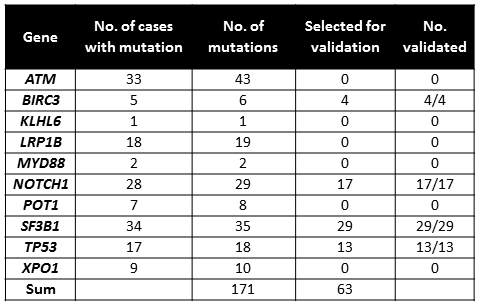
Results: Using the conservative cut-off of 10% for the mutant allele, we found that 105/168 (62.5%) patients carried at least one mutation; ATM (n=33; 20%), BIRC3 (n=5; 3%), NOTCH1 (n=28; 17%), SF3B1 (n=34; 20%) and TP53 (n=17; 10%). Collectively, mutations within these 5 genes accounted for 131/171 (77%) of all mutations observed. Fifty-three patients had more than 1 mutation and 45/53 harbored mutations within more than one gene. We selected 63 mutations for validation, with mutant allele frequencies ranging from 0.10-0.98, and were able to confirm all of them by Sanger sequencing (Table 1).
Summary/Conclusion: With the prognosis of CLL becoming increasingly dependent on an understanding of the molecular landscape of the disease, mutational screening within clinical practice will soon become routine. However, given the large number of potentially significant genetic mutations increasingly being identified, it is apparent that a high-throughput tailored approach is vital in order to efficiently acquire and utilize this data. This study demonstrates the applicability of targeted NGS as a new approach for mutational screening within clinical routine. In the future the need to confirm NGS results by Sanger sequencing will decrease dramatically thereby significantly reducing the amount of labor and costs for clinical laboratories wishing to use NGS technologies.
Keywords: None
Datum přednesení příspěvku: 14. 6. 2014